Facharzt des Artikels

Neue Veröffentlichungen
Hamartom
Zuletzt überprüft: 29.06.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
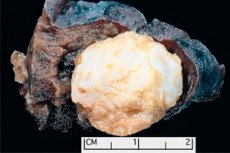
Als Hamartom (vom griechischen hamartia – Fehler, Defekt) wird in der Medizin eine tumorähnliche Formation bezeichnet, die sich in einer beliebigen anatomischen Region befindet und aus dem abnormalen Wachstum von gutartigem Gewebe resultiert. [ 1 ]
Epidemiologie
Statistisch gesehen machen Hamartome 1,2 % der gutartigen Neubildungen aus. Die Prävalenz pulmonaler Hamartome wird auf etwa 0,25 % der Gesamtbevölkerung geschätzt und macht bis zu 8 % aller pulmonalen Neoplasien aus. Die meisten pulmonalen Hamartome werden zufällig bei Patienten im Alter von 40 bis 70 Jahren diagnostiziert, sind in der pädiatrischen Praxis jedoch sehr selten.
Im Allgemeinen werden die meisten Hamartome bei Männern diagnostiziert, obwohl sie in der Niere häufiger bei Frauen vorkommen und im mittleren Alter festgestellt werden.
Etwa 5 % der gutartigen Brusttumore sind Hamartome und betreffen am häufigsten Frauen über 35 Jahren.
80–90 % der hamartomatösen Läsionen des Gehirns und mehr als 50 % der Hamartome des Herzens stehen mit tuberöser Sklerose im Zusammenhang.
Ursachen Hamartome
Gamartome gehören zu den angeborenen Fehlbildungen und sind gutartige Gebilde, die aus mesenchymalen Geweben gebildet werden, die aus Keimblättern stammen. Die Ursachen ihres Auftretens hängen mit der unkontrollierten Zellteilung von zytologisch normalen Geweben (Bindegewebe, glatte Muskulatur, Fettgewebe oder Knorpelgewebe) zusammen, die für eine bestimmte anatomische Stelle charakteristisch sind, und ihrem fokalen Überwachsen während der Embryogenese fast jedes Organs oder jeder anatomischen Struktur.
Das Auftreten mehrerer Hamartome bei demselben Patienten wird oft als Hamartomatose oder pleiotropes Hamartom bezeichnet.
Diese Tumoren können sporadisch oder im Rahmen bestimmter autosomal-dominant vererbter Erkrankungen sowie genetisch bedingter Syndrome auftreten.
In vielen Fällen entstehen Hamartome, wenn sich kurz nach der Geburt eine seltene genetische Erkrankung mit multisystemischer Natur - die tuberöse Sklerose - manifestiert, oder bei der familiären Recklinghausen-Krankheit - der Neurofibromatose Typ 1. [ 2 ]
Risikofaktoren
Zu den Hauptrisikofaktoren für die Bildung eines Hamartoms gehört das Vorhandensein sogenannter genetischer Syndrome der hamartomatösen Polyposis in der Krankengeschichte, darunter:
- Multiples Hamartom-Syndrom – Cowden-Syndrom, bei dem sich multiple Hamartome ekto-, ento- und mesodermalen Ursprungs bilden, gastrointestinale Polypen und mukokutane Manifestationen beobachtet werden;
- Peutz-Jeghers-Turen-Syndrom (gekennzeichnet durch die Entwicklung gutartiger hamartomatöser Polypen im Magen-Darm-Trakt);
- Proteus-Syndrom;
- Weil-Syndrom - juvenile Polyposis des Dickdarms;
- Bannayan-Riley-Ruvalcaba-Syndrom, das wie das Cowden-Syndrom mehrere Hamartome (hamartomatöse Polypen) des Darms verursacht;
- Carney-Stratakis-Syndrom und Carney-Komplex.
Darüber hinaus bilden sich Hamartome bei Patienten mit hereditärem Watson-Syndrom sowie bei sporadisch auftretendem oder angeborenem Pallister-Hall-Syndrom mit hypothalamischem Hamartom und Polydaktylie.
Pathogenese
Der Mechanismus der vermehrten Vermehrung von Keimgewebe mit der Entstehung tumorartiger Fehlbildungen in verschiedenen Organen wird durch Chromosomenaberrationen und Genmutationen erklärt, die spontan auftreten oder vererbt werden können.
Bei der tuberösen Sklerose wurden Mutationen in den Genen TSC1 oder TSC2 identifiziert – Tumorsuppressoren, die eine übermäßige Proliferation – zu schnelles oder unkontrolliertes Zellwachstum und Zellteilung – verhindern und hemmen. Und bei der Neurofibromatose Typ 1 und dem Watson-Syndrom – Keimbahnmutationen des mitochondrialen Tumorsuppressorgens NF1.
Beim Hamartom-Tumorsyndrom, das die Syndrome Cowden, Protea, Bannayan-Riley-Ruvalcaba und juvenile Polyposis kombiniert, ist die Pathogenese mit einer Mutation des PTEN-Gens verbunden, das ein Enzym kodiert, das an der Regulierung der Proliferation beteiligt ist und als Tumorsuppressorgen gilt.
Mutationen im STK11-Gen, das die Struktur und Funktion eines transmembranären Serinenzyms kodiert und dessen Fähigkeit zur Zellteilungseinschränkung reduziert, führen zum Peutz-Jeghers-Turen-Syndrom mit der Entwicklung von Darmpolypen und pigmentierten Hautläsionen. Beim Pallister-Hall-Syndrom wurde eine Mutation im GLI3-Gen identifiziert, einem Transkriptionsfaktor, der an der intrauterinen Gewebebildung beteiligt ist.
Somit führt unkontrolliertes Zellwachstum aufgrund von Genmutationen zur Bildung von Hamartomen.
Symptome Hamartome
Je nach Lokalisation der Hamartome werden ihre Typen unterschieden, und jeder von ihnen hat seine eigene Struktur und Symptomatologie.
Hamartom der Lunge
Ein pulmonales Hamartom kann sich in jedem Lungenlappen und in peripheren Teilen der Lunge bilden und besteht aus normalem Lungengewebe: Fettgewebe, Epithelgewebe, Fasergewebe und Knorpelgewebe. In 80 % der Fälle überwiegt die chondroide Komponente (hyaline Knorpelzellen) unter Einschluss von Adipozyten – Fettgewebezellen und Atemwegsepithelzellen. [ 3 ]
Die früheren Bezeichnungen Chondroides Hamartom, Mesenchymom, chondromatöses Hamartom oder Hamartochondrom werden derzeit von der WHO nicht empfohlen.
Das mesenchymale zystische Hamartom der Lunge kommt dagegen seltener vor und ist bei den meisten Patienten mit dem Cowden-Syndrom assoziiert.
Eine hamartomatöse Läsion der Lunge kann sich zwar nicht manifestieren, kann aber Symptome in Form von chronischem Husten (oft mit Hämoptyse), Keuchen beim Atmen und Atembeschwerden verursachen. [ 4 ]
Ein Hamartom des Herzens
Zu den gutartigen primären Herztumoren zählen bei Erwachsenen das Hamartom reifer Myozyten und bei Säuglingen und Kindern mit tuberöser Sklerose das Rhabdomyom, d. h. das myokardiale Hamartom der Ventrikel oder des interventrikulären Septums. [ 5 ]
Ein reifes Kardiomyozytenhamartom entwickelt sich in der Ventrikelwand (seltener auch in den Vorhöfen) und kann als multiple, dichte Massen auftreten, die eng mit dem darunterliegenden Myokard verbunden sind. Der Tumor kann Symptome einer Herzinsuffizienz verursachen: Brustschmerzen, Herzklopfen und Herzrhythmusstörungen, Herzgeräusche, Ödeme, Dyspnoe und Zyanose.
Kardiale Rhabdomyome werden meist im ersten Lebensjahr diagnostiziert. Sie bestehen aus Herzmuskelgewebe, das von embryonalen Myoblasten gebildet wird, und haben das Aussehen fester, fokaler Massen ohne Kapsel.
Typischerweise treten diese Hamartome asymptomatisch auf und bilden sich vor dem vierten Lebensjahr spontan zurück.
Einige Experten gehen auch davon aus, dass hamartomatöse Läsionen mit dem Carney-Komplex -Myxom des Herzens in Zusammenhang stehen. [ 6 ]
Hamartom des Gastrointestinaltrakts
Das Magenhamartom ist eine mesenchymale Raumforderung in Form eines epithelialen hyperplastischen Polypen des Magens, eines Peutz-Jeghers-Polypen und eines seltenen myoepithelialen Hamartoms – mit hypertrophierten glatten Muskelbündeln. Andere Bezeichnungen für dieses Hamartom sind myoglanduläres Hamartom, adenomyomatöses Hamartom und Magenadenomyom. Typische klinische Manifestationen sind Dyspepsie, epigastrische Schmerzen und Blutungen im oberen Gastrointestinaltrakt. [ 7 ], [ 8 ]
Weitere Informationen im Material - Magenpolyposis
Ein intestinales Hamartom ist ein hamartomatöser oder hyperplastischer Polyp des Dickdarms, der als adenomatöses oder tubuläres Adenom diagnostiziert wird. Befindet sich das Hamartom in der Brunner-Drüse des Duodenums, manifestieren sich die Symptome durch Schmerzen im Oberbauch, Übelkeit, Erbrechen und Blähungen (Hinweis auf einen Darmverschluss) sowie bei erheblicher Größe durch gastrointestinale Blutungen. Bei einem myoepithelialen Hamartom des Ileums klagen die Patienten über Bauchschmerzen, Gewichtsverlust und entwickeln eine chronische Anämie. [ 9 ], [ 10 ]
Lesen Sie auch – Rektumpolypen
Das retrorektale Hamartom ist ein zystisches Hamartom oder eine mehrkammerige Zyste des retrorektalen Raums (des lockeren Bindegewebes zwischen Rektum und seiner eigenen Faszie), die am häufigsten bei Frauen mittleren Alters auftritt. Es sieht aus wie eine aus der mit Epithel ausgekleideten Rektumrückwand hervortretende Zyste mit chaotisch angeordneten glatten Muskelfasern. Dieses Hamartom äußert sich in Unterleibsschmerzen und wiederkehrender Verstopfung. [ 11 ], [ 12 ]
Hamartome der Leber und Milz
Das multiple biliäre Hamartom der Leber ist ein Hamartom der interdolischen intrahepatischen Gallengänge, das mit Fehlbildungen ihrer Entwicklung während der Embryonalperiode einhergeht. Dieses Hamartom (einzeln oder multipel) besteht aus zufällig erweiterten Ansammlungen von Gallengängen und fibrokollagenem Stroma. [ 13 ]
Gallenhamartome sind asymptomatisch und werden in der Regel zufällig (bei einer radiologischen Untersuchung oder Laparotomie) entdeckt. [ 14 ]
Eine seltene und oft zufällig entdeckte primäre Neoplasie benignen Charakters ist ein Hamartom der Milz, das aus Elementen der roten Milzpulpa besteht - in Form einer gut abgegrenzten homogenen Masse von fester Konsistenz. Diese Fehlbildung kann einzeln oder mehrfach auftreten; beim Zusammendrücken des Milzparenchyms kann es zu Beschwerden und Schmerzen im linken subkostalen Bereich kommen. [ 15 ], [ 16 ]
Nierenhamartome
Das häufigste Hamartom der Niere wird als Angiomyolipom der Niere diagnostiziert, da dieser gutartige Tumor aus reifem Fettgewebe mit eingebetteten glatten Muskelfasern und Blutgefäßen besteht. Er entsteht bei tuberöser Sklerose in 40-80 % der Fälle. Eine Vergrößerung des Hamartoms (mehr als 4-5 cm) führt zu Schmerzen und Blut im Urin. [ 17 ], [ 18 ]
Hamartom der Brust
Die von der WHO anerkannten diagnostischen Definitionen des Brusthamartoms sind Begriffe wie Adenolipom, Chondrolipom und myoides Hamartom. Obwohl es von Mammologen oft als Fibroadenolipom bezeichnet wird, besteht die Tumorformation aus Zellen des Binde-, Drüsen- und Fettgewebes, die von einer dünnen, klar umrissenen Bindegewebskapsel umgeben sind. Bei der Visualisierung können fokale Verkalkungen beobachtet werden. In diesem Fall fehlen klinische Manifestationen. [ 19 ], [ 20 ]
Lesen Sie auch — Brusttumore
Hamartome des Gehirns
Ein Drittel der Patienten mit tuberöser Sklerose weist ein Hirnhamartom in Form intrakranieller kortikaler Auswüchse oder Tuberkel in verschiedenen Hirnlappen – an der Grenze zwischen grauer und weißer Substanz – oder subependymaler Knötchen entlang der Wände der Hirnventrikel auf. Auch ein astrozytisches Hamartom, ein subependymales Riesenzellastrozytom mit kortikaler Störung, dysmorphen Neuronen und großen Gliazellen des Hirnparenchyms (Astrozyten), kann sich bilden. Zu den Symptomen zerebraler Hamartome gehören Krampfanfälle und geistige Behinderung bei Kindern. [ 21 ], [ 22 ]
Eine seltene Fehlbildung, die während der Embryogenese auftritt und bei der Geburt vorhanden ist, ist ein hypothalamisches Hamartom, eine Ansammlung heterotoper Neuronen und Gliazellen. Mit dem Wachstum des kindlichen Gehirns vergrößert sich der Tumor, breitet sich aber nicht auf andere Hirnregionen aus. [ 23 ], [ 24 ]
Wenn sich im vorderen Teil des Hypothalamus (Tuber cinereum), wo die Hypophyse ansetzt, hypertrophiertes Gewebe bildet, manifestiert sich die Fehlbildung in Symptomen einer zentralen vorzeitigen sexuellen Entwicklung (vor dem 8.-9. Lebensjahr): Auftreten von Akneausschlägen, frühe Entwicklung der Brustdrüsen und frühe Menarche bei Mädchen; frühe Schambehaarung und Stimmveränderung bei Jungen.
Wenn sich im hinteren Teil des Hypothalamus Hamartome bilden, kann es zu Anomalien der elektrischen Aktivität des Gehirns kommen, die sich im frühen Säuglingsalter durch Krampfanfälle und in einem späteren Stadium (im Alter von 4 bis 7 Jahren) durch Epilepsie mit fokalen epileptischen Anfällen mit plötzlichem Lachen oder mit unwillkürlichem Weinen, atonischen und tonisch-klonischen Anfällen sowie Aggressionsanfällen, Gedächtnis- und kognitiven Problemen äußern.
Ein Hypophysenhamartom ist ein sporadisch auftretendes gutartiges Hypophysenadenom.
Bei Erwachsenen mittleren Alters mit Cowden-Syndrom kann eine seltene tumorartige Masse, ein Hamartom des Kleinhirns, auftreten, die als dysplastisches Kleinhirngangliozytom oder Lhermitte-Duclos-Syndrom diagnostiziert wird. Symptome können fehlen oder sich als Kopfschmerzen, Schwindel, beeinträchtigte Bewegungskoordination und Lähmung einzelner Hirnnerven äußern.
Lymphknotenhamartom
Wenn die Zellen der glatten Muskulatur und des Fettgewebes sowie die Blutgefäße und das kollagene Stroma der inguinalen, retroperitonealen, submandibulären und zervikalen Lymphknoten überwachsen, entsteht ein angiomyomatöses Hamartom eines Lymphknotens oder ein noduläres angiomyomatöses Hamartom - mit teilweisem oder vollständigem Ersatz seines Parenchyms. [ 25 ], [ 26 ]
Ein Hamartom der Haut
Bei Vorliegen einer tuberösen Sklerose oder Neurofibromatose werden verschiedene Hamartome der Haut beobachtet, am häufigsten in Form von hypopigmentierten Flecken; Kaffee- und Milchflecken; Angiofibrom (an den Wangen, am Kinn, in den Nasolabialfalten); Chagrin-Flecken verschiedener Lokalisationen (das sind Bindegewebsnävi); faserige Plaques an der Stirn, der Kopfhaut oder am Hals.
Eine seltene dermatologische Manifestation der tuberösen Sklerose (vor allem bei Männern) ist das follikulozystische und kollagene Hamartom, das durch reichliche Kollagenablagerung in der Dermis, konzentrische perifollikuläre Fibrose und keratingefüllte trichterförmige subkutane Zysten gekennzeichnet ist, die bei der histopathologischen Untersuchung sichtbar werden. [ 27 ]
Zu den aus Melanozyten (Zellen, die das Pigment Melanin produzieren) bestehenden Hamartomen zählen die meisten Experten auch verschiedene melanozytäre Neoplasien, insbesondere kongenitale melanozytäre Nävi, die eine Anomalie der Embryogenese darstellen.
Ätiologiemäßig handelt es sich bei den aus Gefäßgewebe bestehenden Hamartomen ebenfalls um Hämangiome der Haut.
Patienten mit Peutz-Jeghers-Thuren-Syndrom haben ein Hamartom in Form einer fleckigen Pigmentierung der Haut und Schleimhäute - Lentiginosis periorificialis
Fälle von linearem papulösem ektodermal-mesodermalem Hamartom (Hamartoma moniliformis) zeigen einen linearen fleischfarbenen papulösen Ausschlag an Kopf, Hals und oberer Brust.
Und ein sebozytisches Hamartom ist ein Hamartom der Talgdrüsen, lesen Sie mehr in der Veröffentlichung – Talgdrüsennävus.
Hamartom des Auges
Pigmentierte hamartomatöse Läsionen der Iris bei Neurofibromatose Typ 1 und Watson-Syndrom – in Form von knotigen Ansammlungen dendritischer Melanozyten – werden als Irishamartome oder Lisch-Knoten bezeichnet. Es handelt sich um transparente (normalerweise das Sehvermögen nicht beeinträchtigende), abgerundete, kuppelförmige gelbbraune Papeln, die über die Oberfläche der Iris hinausragen.
Und Patienten mit juvenilem Angiofibrom des Nasopharynx und familiärer adenomatöser Polyposis entwickeln häufig ein kombiniertes Hamartom der Netzhaut und des retinalen Pigmentepithels – in Form eines schwarzen Flecks im zentralen (makulären) Teil der Netzhaut. [ 28 ]
Ein Hamartom der Nase
Das nasale Hamartom wird von Spezialisten als nasales Chondromesenchymales Hamartom oder nasales Chondrom definiert und ist auf eine gutartige Proliferation des respiratorischen Epithels, der submukösen Drüsen und des Chondroknochenmesenchyms zurückzuführen. Die klinischen Manifestationen hängen von Größe und Lokalisation der Läsion ab und umfassen: verstopfte Nase, Schwierigkeiten bei der Nasenatmung und beim Stillen bei Säuglingen, klaren, wässrigen Nasenausfluss und Nasenbluten. Ein Hamartom kann mit dem Kind wachsen und sich in die Augenhöhlen ausbreiten, was zu einer Vorwärts- oder Rückwärtsverschiebung des Augapfels, Strabismus oder Okulomotorikstörungen führen kann. [ 29 ]
Ein Hamartom bei einem Kind
Alle oben genannten hamartomatösen Läsionen verschiedener Organe und anatomischer Strukturen sind bei Kindern mit entsprechenden Syndromen vorhanden.
Neugeborene weisen ein mesenchymales Hamartom der Brustwand oder ein knorpeliges Hamartom der Rippen auf. Dabei handelt es sich um feste, unbewegliche Massen, die durch ein fokales Überwuchern normaler Skelettelemente mit knorpeligen, vaskulären und mesenchymalen Elementen entstehen. Dieses Hamartom kann zu Atemversagen und der Entwicklung eines Atemnotsyndroms führen. Das mesenchymale Hamartom der Leber ist der zweithäufigste gutartige Lebertumor bei Kindern. Diese tumorartige Formation (häufiger im rechten Lappen des Organs lokalisiert) besteht aus Zellen des mesenchymalen Stromas, Hepatozyten und Epithelzellen der Gallengangsauskleidung. Das klinische Bild umfasst eine tastbare Masse in der Bauchhöhle, Anorexie und Gewichtsverlust. Bei erheblicher Größe (bis zu 10 cm und mehr) bedeckt der Tumor die extrahepatischen Gallengänge und die untere Hohlvene, was zu Gelbsucht und Ödemen der unteren Extremitäten führt.
Ein Hamartom ist ein angeborenes mesoblastisches Nephrom (tritt bei 1 von 200.000 Säuglingen auf), das bei Neugeborenen zu Blähungen im Bauchraum mit einer tastbaren Masse von dichter Konsistenz im rechten Oberbauch führen kann. Säuglinge können auch eine schnelle, flache Atmung aufweisen.
Zu den seltenen angeborenen Anomalien gehört das fibröse Hamartom des Säuglingsalters, das bei Kindern in den ersten beiden Lebensjahren auftritt und sich als schmerzlose knotige Masse im Unterhautgewebe der Achselhöhle, des Halses, der Schulter und des Unterarms, des Rückens und der Brust, des Oberschenkels, des Fußes und der äußeren Genitalien präsentiert.
Ein ekkrines angiomatöses Hamartom kann bei Kindern bereits bei der Geburt vorhanden sein oder sich bereits in der frühen Kindheit manifestieren. Dieser gutartige Tumor hamartomatöser Natur zeigt in der Regel bläuliche oder bräunliche Knötchen und/oder Plaques, die durch die Proliferation von ekkrinem Schweißdrüsengewebe und Kapillaren in den mittleren und tiefen Schichten der Dermis entstehen. Dieses Hamartom kann zu lokaler Hyperhidrose und verstärktem Haarwachstum führen.
Komplikationen und Konsequenzen
Es besteht allgemein Einigkeit darüber, dass Hamartome selten wiederkehren oder sich in bösartige Tumoren verwandeln. Sie zeigen oft kaum oder gar keine Symptome und verschwinden manchmal sogar mit der Zeit. In schwereren Fällen und je nach Entstehungsort können diese Missbildungen jedoch schwerwiegende Komplikationen und Folgen haben.
Zunächst einmal kann ein Hamartom eine solche Größe erreichen, dass es auf umliegendes Gewebe und Organe drückt und deren Funktionen beeinträchtigt.
Ein kardiales Hamartom kann bei Kindern zu anhaltenden Herzrhythmusstörungen, Klappendefekten und einer Beeinträchtigung des intrakardialen Blutflusses mit nachfolgender Herzinsuffizienz führen.
Komplikationen hamartomatöser Polypen des Gastrointestinaltrakts sind gastrointestinale Blutungen, Obstruktionen und Darmintussuszeption (mit möglichem tödlichem Ausgang). Ein großes Nierenhamartom kann zudem eine Nierenruptur hervorrufen.
Ein Hamartom im Gehirn kann ein obstruktives Hydrozephalus-Syndrom verursachen.
Bei hypothalamischen und hypophysären Hamartomen kann die Produktion des somatotropen Hormons (Wachstumshormon) beeinträchtigt sein, was bei Kindern zur Entwicklung eines Hypophysenmangels (Hypopituitarismus) führen kann. Hypothalamische Hamartome können bei Kindern auch zu medikamentenresistenter Epilepsie führen.
Zu den Komplikationen des Hamartoms des retinalen Pigmentepithels zählen Funktionsstörungen der Netzhaut und/oder des Sehnervs, ein Makulaödem, eine Neovaskularisierung der Aderhaut und eine Netzhautablösung.
Diagnose Hamartome
Ein wichtiger Bestandteil der Diagnose von Hamartomen und verwandten Syndromen ist die Erhebung der Anamnese, einschließlich der Familienanamnese.
Zu den Laboruntersuchungen gehören Blutuntersuchungen: allgemeine klinische Daten, Serumelektrolyte, Lymphozytenprofil, Kalzium-, Kalium-, Phosphat- und Harnstoffwerte sowie Leberfunktionstests. Wenn möglich, wird eine Feinnadelaspirationspunktion der Masse durchgeführt, da die histologische Untersuchung für die Diagnose und die Wahl der Behandlungstaktik entscheidend ist.
Die instrumentelle Diagnostik ermöglicht die Visualisierung hamartomatöser tumorartiger Gebilde und die Identifizierung ihrer genauen Lokalisation. Hierzu werden Röntgen, Angiographie, Elektroenzephalographie (EEG), Ultraschall (Sonographie), CT (Computertomographie), PET (Positronen-Emissions-Tomographie) und MRI (Magnetresonanztomographie) eingesetzt.
Differenzialdiagnose
Bei allen auffälligen Tumoren ist die Differenzialdiagnose von großer Bedeutung. So werden Tuberkulom und Hamartom unterschieden; pulmonales Hamartom und primärer Lungenkrebs, bronchogenes Karzinoid und metastasierende Erkrankung. Das Hirnhamartom muss vom Kraniopharyngeom und dem hypothalamisch-chiasmatischen Gliom unterschieden werden. Die Differenzialdiagnose des Hamartoms als kongenitales mesoblastisches Nephrom umfasst den Wilms-Tumor (malignes Nephroblastom), das klarzellige Sarkom der Niere und den ossifizierenden Nierentumor bei Säuglingen.
Wen kann ich kontaktieren?
Behandlung Hamartome
Wenn das Hamartom asymptomatisch ist und zufällig entdeckt wird, ist keine Behandlung erforderlich. Es ist jedoch notwendig, sein „Verhalten“ und den Zustand des Patienten zu überwachen. In anderen Fällen zielt die Therapie darauf ab, die Intensität der Symptome zu reduzieren und Komplikationen vorzubeugen. Beispielsweise werden bei einem hypothalamischen Hamartom mit Symptomen einer vorzeitigen Pubertät bestimmte Medikamente verschrieben, die die Freisetzung bestimmter Hormone hemmen. Herzmedikamente werden zur Behandlung von Symptomen einer Herzinsuffizienz bei Patienten mit Herzhamartomen eingesetzt.
Die operative Entfernung von Hamartomen ist zur Diagnosesicherung und bei medizinisch nicht behandelbaren intensiven Beschwerden angezeigt.
Beispielsweise können Lungenhamartome durch eine Keilresektion und in schweren Fällen durch die Entfernung eines Lungenlappens (Lobektomie) entfernt werden. Auch ein Brusthamartom kann exzidiert werden. Bei großen Ausmaßen kann eine teilweise oder vollständige Mastektomie erforderlich sein.
Zur Entfernung hamartomatöser Polypen können stereotaktische Radiofrequenz-Thermoablation oder Laserablation eingesetzt werden. Auch die Radiochirurgie mit hochfokussierten Gammastrahlen – Gamma-Knife bei hypothalamischen Hamartomen oder astrozytären Hamartomen – kommt zum Einsatz.
Verhütung
Die einzige Möglichkeit, die Entwicklung von Hamartomen zu verhindern, kann ein genetisches Screening der zukünftigen Eltern des Kindes sein.
Prognose
Die Gesamtprognose dieser angeborenen Anomalie hängt von der Lokalisation und Größe des Neoplasmas sowie von Begleiterkrankungen und dem allgemeinen Gesundheitszustand des Patienten ab.