Facharzt des Artikels

Neue Veröffentlichungen
Choanal-Atresie
Zuletzt überprüft: 29.06.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
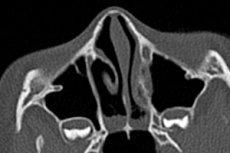
Das völlige Fehlen natürlicher Öffnungen in verschiedenen anatomischen Strukturen des Körpers wird als Atresie bezeichnet (von griechisch a – Verleugnung von etwas, tresis – Öffnung). Choanalatresie bezeichnet das Fehlen paariger Öffnungen im hinteren Teil des Nasengangs – den hinteren Nasengängen, die die Nasenhöhle mit dem Nasenrachen verbinden. [ 1 ]
Epidemiologie
Die Häufigkeit dieser Fehlbildung beträgt einen Fall pro 5.000 bis 8.000 Lebendgeburten (anderen Angaben zufolge drei Fälle pro 10.000), und in 65 % der Fälle werden Kinder mit einseitiger Choanalatresie geboren.
Gleichzeitig weisen Neugeborene in 60–75 % der Fälle bilateraler Atresie begleitende Fehlbildungen auf – andere kraniofaziale Anomalien. Darüber hinaus verzeichnet die Statistik fast 8 % familiäre Fälle.
Einigen Berichten zufolge weisen bis zu 30 % der Patienten beim CHARGE-Syndrom eine Choanalatresie auf. [ 2 ]
Ursachen Choanalatresie
Da es sich bei der Choa-Atresie bei Neugeborenen um eine angeborene Erkrankung handelt, liegen ihre Ursachen in der Störung der Nasenstrukturen während der embryonalen Entwicklung. Infolge dieser Störungen verbleibt zwischen der Nasenhöhle (Cavum nasi) und dem oberen Teil des Nasenrachens (Pars nasalis pharyngis) ein knöchernes/knorpeliges Septum oder, seltener, eine fibröse (Bindegewebe-)Membran.
Der genetische Faktor sollte berücksichtigt werden, insbesondere wenn ein Komplex von Entwicklungsdefekten vorliegt, wie beispielsweise das angeborene CHARGE-Syndrom oder die cHARGE-Assoziation – mit Anomalien der Augenmembranen, Ohrmuscheln, Speiseröhre, Genitalien usw. Zu den angeborenen Syndromen der kraniofazialen (kraniofazialen) Dysostose oder Kraniosyntose (vorzeitige Fusion einer oder mehrerer Schädelnähte), die durch Genmutationen verursacht werden und bei denen eine Anomalie des Nasopharynx und der hinteren Nasengänge vorliegt, gehören auch das Treacher-Collins-Syndrom; Alfie-, DiGeorgi-, Apert-Syndrom; Edwards-Syndrom; Crouzon-, Antley-Bixler-, Pfeiffer-, Tessier-, Beer-Stevenson-, Jackson-Weiss-Syndrom; das fetale Alkoholsyndrom (fetales Alkoholsyndrom).
Bei der Entwicklung einer deformierenden Nasenpolyposis bei Kindern und jungen Erwachsenen liegt eine Choanalstenose vor, also eine krankhafte Verengung, die als Einengung der Nasenwege im hinteren Choanalbereich, Nasenrachenstenose oder partielle Choanalatresie definiert werden kann.
So wird in der HNO-Heilkunde häufig auch eine erworbene Choanalatresie erkannt – eine sekundäre vordere und hintere Stenose der Nasenhöhle mit Bildung fibröser Septen. Dieser Zustand kann die Folge von Syphilis, systemischem Lupus erythematodes, einem Trauma der Nasennebenhöhlen, einem chirurgischen Eingriff sowie eine Folge einer Strahlentherapie bei bösartigen Tumoren des Nasopharynx sein.
Allerdings wird die Choanalatresie von medizinischen Experten als angeborene Pathologie eingestuft und praktizierende HNO-Ärzte sollten sie von einer Stenose der hinteren Nasengänge unterscheiden, die nicht zu einer vollständigen Obstruktion führt.
Doppelt so häufig ist eine unilaterale Atresie: die rechtsseitige Choanalatresie bzw. die linksseitige Choanalatresie. [ 3 ]
Risikofaktoren
Neben genetischen Anomalien gelten verschiedene embryotoxische Belastungen und Umweltfaktoren als Risikofaktoren für eine Atresie des Foramen distal nasalis.
Daher besteht ein höheres Risiko für diese Anomalie beim Fötus bei werdenden Müttern, die während der Schwangerschaft Medikamente der Thioamid-Gruppe gegen Hyperthyreose eingenommen haben (um den Schilddrüsenhormonspiegel zu senken). In solchen Fällen kann es zu einem Mangel an Schilddrüsenhormonen im Embryo kommen, was sich negativ auf die Morphogenese der oberen Atmungsorgane auswirkt.
Darüber hinaus haben Studien einen möglichen Zusammenhang zwischen neonataler Choanalatresie und hohen Dosen von Vitamin B12, B3 (PP), D und Zink während der Schwangerschaft festgestellt. Alkohol, Tabakrauch und Koffein wirken sich äußerst negativ auf die Entwicklung der kraniofazialen Strukturen des Fötus aus. [ 4 ]
In den Jahren 2010 bis 2012 wurde in den USA ein Anstieg der Geburten mit Choanalatresie gemeldet, da schwangere Frauen Chemikalien ausgesetzt waren, die zur Behandlung von Nutzpflanzen verwendet werden.
Mehr lesen:
Pathogenese
Choans (lat. Choane (lat.: Trichter) sind Öffnungen, die von der Nasenhöhle in den Nasenrachenraum führen und in der Mitte durch die Pfanne (Rand der Knochenplatte) begrenzt sind; Keilbein - von oben und hinten; Flügelplatten dieses Knochens - von den Seiten und von unten - Gaumenbein (seine horizontale Platte). Weitere Informationen im Material - Entwicklung der Atmungsorgane
Die Bildung der Choanen, die aus den Kiemenbögen des Embryos stammen, beginnt in der vierten Schwangerschaftswoche (und dauert bis zur achten Woche) mit der Migration von Neuralleistenzellen in die dorsalen Neuralfalten. Anschließend reißt die vertikal angeordnete Epithelfalte (Oronasalmembran) zwischen dem Dach der primären Mundhöhle und den Nasenfortsätzen (Placoda nasalis) an der lateralen Kopffläche. Die Nasenfortsätze vertiefen sich in das Mesoderm, was zur Bildung von Nasengruben und anschließend zu den primären (primitiven) Choanen führt.
Theoretisch könnte die Pathogenese der angeborenen Anomalie der Choanalatresie auf den Erhalt der Wangen-Rachen-Membran (Bukkopharyngealmembran) zurückzuführen sein, einer dünnen Schicht aus Ektoderm- und Entodermzellen, die die Mundöffnung des Embryos oberhalb des kranialen Endes der Chorda bedeckt. Diese Membran sollte in der sechsten Schwangerschaftswoche perforieren, was jedoch aus unbekannten Gründen nicht der Fall sein kann, was zu orofazialen Defekten wie Gaumenspalten und Choanalatresie führt.
Ebenfalls möglich: Erhalt der Bucconasalmembran (eine dünne Schicht Epithelgewebe, die in der siebten Schwangerschaftswoche resorbiert werden sollte); abnorme Anhaftung von mesodermalem Gewebe im Bereich der Choanen; lokale Störung der Migration mesenchymaler Zellen entlang der Neuralleiste, was zu Defekten bei der Bildung der frontonasalen Vorwölbung des Kopfteils des Embryos und seiner Verzweigungen führt.
Für keine der Annahmen zum Entstehungsmechanismus der hinteren Nasenatresie gibt es bisher jedoch Belege.
Symptome Choanalatresie
Neugeborene atmen durch die Nase, da ihr Kehldeckel höher liegt (im Vergleich zu Erwachsenen) und der Kehlkopf beim Schlucken ansteigt, den Nasopharynx berührt und sich zwischen dem weichen Gaumen und den Seiten des Nasopharynx schließt. Die Fähigkeit, durch den Mund zu atmen, tritt 4-6 Wochen nach der Geburt auf – nach der Senkung des Kehlkopfes.
Daher sind die klassischen Symptome einer bilateralen Choanalatresie bei Neugeborenen auf eine vollständige Beeinträchtigung der Atemfunktion zurückzuführen.
Beispielsweise weist das Kind eine zyklische Zyanose auf, die auf Asphyxie-Episoden hindeutet: eine bläuliche Verfärbung der Haut, die beim Weinen (wenn das Kind den Mund weit öffnet und ein- und ausatmet) abnimmt und wiederkehrt, sobald das Weinen aufhört und das Kind den Mund schließt. In solchen Fällen ist eine medizinische Notfallversorgung erforderlich – eine endotracheale Intubation oder Tracheotomie.
Eine unilaterale Atresie (das Fehlen nur eines hinteren Nasengangs) wird oft erst im späteren Lebensalter (im Alter von 5–10 Monaten oder deutlich später) festgestellt. Die ersten Anzeichen sind eine einseitige Verstopfung der Nase. Zusätzlich kommt es zu anhaltendem Ausfluss aus einem Nasenloch ( Rhinorrhoe ), Schnarchen und Stridor (geräuschvolle Atmung) sowie einer chronischen Nasennebenhöhlenentzündung. [ 5 ]
Komplikationen und Konsequenzen
Eine bilaterale Choanalatresie führt aufgrund der vollständigen Obstruktion der Nasenwege zu einem akuten Atemnotsyndrom bei Neugeborenen.
Folgen und Komplikationen einer unilateralen Atresie: Verzerrung der Gesichtsproportionen, gestörtes Wachstum des Ober- und Unterkiefers und Bildung eines pathologischen Bisses – aufgrund einer fehlerhaften kraniofazialen Entwicklung; Auftreten von obstruktiver nächtlicher Apnoe und anderen Atemproblemen, die mit einer Funktionsstörung der oberen Atemwege verbunden sind. [ 6 ]
Diagnose Choanalatresie
Bei Verdacht auf eine bilaterale Choanalatresie bei Neugeborenen wird die vorläufige klinische Diagnose im Notfall von einem Neonatologen durch das Einführen einer Magensonde durch die Nasenhöhle des Säuglings gestellt. Der Verdacht auf diese angeborene Anomalie bestätigt sich, wenn der Katheter nicht eingeführt werden kann.
Zur Sicherung der Diagnose sind bildgebende Verfahren notwendig: Endoskopie (Untersuchung) der Nasenhöhle, Computertomographie (CT) der Nase, der Nasennebenhöhlen und der Nasennebenhöhlenknochenstrukturen.
Die am häufigsten auftretende Form ist die einseitige Choanalatresie. Sie geht möglicherweise nicht mit Geburtsfehlern einher und wird daher möglicherweise nicht unmittelbar nach der Geburt diagnostiziert.
Bei einseitiger Atresie wird auch eine instrumentelle Diagnostik durchgeführt: vordere und hintere Rhinoskopie; Endoskopie der Nasenhöhle und nasale Computertomographie; Rhinomanometrie - Untersuchung der nasalen Atmungsfunktion.
Differenzialdiagnose
Die Differentialdiagnose umfasst Nasenatmungsprobleme, die folgende Ursachen haben können: Nasenscheidewandverkrümmung oder Knorpelverrenkung; Nasenhöhlenstenose und angeborene Hypertrophie der unteren Nasenbeine; isolierte Stenose des birnenförmigen Nasenlochs (vordere knöcherne Einengung des Nasenskeletts); Anthrochoanalpolyp, Dermoidzyste der Nasenhöhle oder Tränennasengangzyste; Hämangiom oder Nasengarmatom.
Wen kann ich kontaktieren?
Behandlung Choanalatresie
Bei einer Choanalatresie kann zur Wiederherstellung der Durchgängigkeit nur eine chirurgische Behandlung mittels transnasaler endoskopischer Resektion und Choanoplastik mit vorheriger CT- oder MRT-Untersuchung der Nasenhöhle durchgeführt werden.
Der chirurgische Eingriff bei beidseitiger Choanalatresie erfolgt in der Regel innerhalb der ersten drei Lebensmonate, bei einseitiger Atresie nach dem zweiten Lebensjahr des Kindes. [ 7 ]
Alle Details in der Publikation - Rekonstruktion der Choanalatresie
Verhütung
Angesichts der bekannten Risikofaktoren für diesen Geburtsfehler können vorbeugende Maßnahmen in einer angemessenen Schwangerschaftsbegleitung und äußerster Vorsicht bei der Verschreibung von Medikamenten an werdende Mütter bestehen.
Und verantwortungsbewusste Paare können der Geburt eines Kindes mit einem genetisch bedingten Syndrom vorbeugen, beispielsweise durch eine medizinisch-genetische Beratung.
Prognose
Eine bilaterale Choanalatresie ist für das Neugeborene lebensbedrohlich, doch bei rechtzeitiger und wirksamer Behandlung und ohne Zusammenhang mit angeborenen Syndromen ist die Prognose im Allgemeinen gut.
Bücher über Atresie der Choanen
- „Pediatric Otolaryngology: Principles and Practice Pathways“ – von Christopher J. Hartnick et al. (Erscheinungsjahr: 2015)
- „Scott-Brown's Otorhinolaryngology, Head and Neck Surgery“ – Autor: John C Watkinson et al. (Erscheinungsjahr: 2020)
- „Cummings Otolaryngology: Head and Neck Surgery“ – Autor: Paul W. Flint et al. (Erscheinungsjahr: 2020)
- „HNO: Eine Einführung und ein praktischer Leitfaden“ – Autorin: Sharan K. Naidoo (Erscheinungsjahr: 2018)
Verwendete Literatur
- Palchun, Magomedov, Alexeeva: Hals-Nasen-Ohrenheilkunde. Nationales Handbuch. GEOTAR-Medien, 2022.
- Angeborene Choanalatresie bei Kindern. Lehrbuch für Medizinstudenten. Kotova EN, Radtsig E.Yu. 2021