Facharzt des Artikels

Neue Veröffentlichungen
Treacher-Collins-Syndrom
Last reviewed: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.

Intrauterine Störungen der Knochenentwicklung führen zu schweren kraniofazialen Deformitäten, und eine der Varianten dieser Pathologie ist das Treacher-Collins-Syndrom (TCS) oder die mandibulofasziale, d. h. maxillofaziale Dysostose.
Krankheitscode nach ICD 10: Klasse XVII (angeborene Anomalien, Deformationen und Chromosomenstörungen), Q75.4 – Mandibulofaziale Dysostose.
Ursachen Treacher-Collins-Syndrom
Dieses Syndrom wurde nach dem herausragenden britischen Augenarzt Edward Treacher Collins benannt, der die Hauptmerkmale der Pathologie vor über hundert Jahren beschrieb. Europäische Ärzte bezeichnen diese Art von Gesichts- und Kieferknochenanomalie jedoch häufiger als Franceschetti-Syndrom – basierend auf den umfangreichen Forschungen des Schweizer Augenarztes Adolf Franceschetti, der Mitte des letzten Jahrhunderts den Begriff „Mandibulofasziale Dysostose“ prägte. In medizinischen Kreisen wird auch der Name Franceschetti-Collins-Syndrom verwendet.
Das Treacher-Collins-Syndrom wird durch Mutationen im TCOF1-Gen (am Chromosomenort 5q31.3-33.3) verursacht, das für ein nukleolares Phosphoprotein kodiert, das für die Bildung des kraniofazialen Teils des menschlichen Embryos verantwortlich ist. Durch einen vorzeitigen Rückgang dieses Proteins werden die Biogenese und die Funktionen der rRNA gestört. Laut Genetikern des Humangenomforschungsprogramms führen diese Prozesse zu einer verminderten Proliferation embryonaler Zellen der Neuralleiste – einem Grat entlang der Neuralrinne, der sich während der Embryonalentwicklung zu einem Neuralrohr verschließt.
Die Bildung von Gesichtsgewebe erfolgt durch Transformation und Differenzierung von Zellen des oberen (Kopf-)Teils der Neuralleiste, die entlang des Neuralrohrs in den Bereich des ersten und zweiten Kiemenbogens des Embryos wandern. Und der Mangel dieser Zellen verursacht kraniofaziale Deformationen. Der kritische Zeitraum für das Auftreten von Anomalien liegt zwischen 18 und 28 Tagen nach der Befruchtung. Nach Abschluss der Migration der Neuralleistenzellen (in der vierten Schwangerschaftswoche) sind fast alle lockeren mesenchymalen Gewebe im Gesichtsbereich gebildet, die sich später (von der 5. bis zur 8. Woche) in Skelett- und Bindegewebe aller Teile des Gesichts, des Halses, des Kehlkopfes, des Ohrs (einschließlich des Innenohrs) und der zukünftigen Zähne differenzieren.
Pathogenese
Die Pathogenese des Treacher-Collins-Syndroms ist häufig familiär bedingt und die Anomalie wird autosomal-dominant vererbt, obwohl es Fälle einer autosomal-rezessiven Vererbung des Defekts (mit Mutationen in anderen Genen, insbesondere POLR1C und POLR1D) gibt. Das Unvorhersehbarste an der maxillofazialen Dysostose ist, dass die Mutation nur in 40-48 % der Fälle an Kinder vererbt wird. Das heißt, bei 52-60 % der Patienten sind die Ursachen des Treacher-Collins-Syndroms nicht mit dem Vorhandensein einer Anomalie in der Familie verbunden, und es wird angenommen, dass die Pathologie als Folge sporadischer Genmutationen de novo auftritt. Neumutationen sind höchstwahrscheinlich die Folgen teratogener Effekte auf den Fötus während der Schwangerschaft.
Zu den teratogenen Ursachen dieses Syndroms zählen Experten hohe Dosen von Ethanol (Ethylalkohol), Strahlung, Zigarettenrauch, Cytomegavirus und Toxoplasma sowie Herbizide auf Glyphosatbasis (Roundal, Glyfor, Tornado usw.). Die Liste der iatrogenen Faktoren umfasst Akne- und Seborrhoe-Medikamente mit 13-cis-Retinsäure (Isotretinoin, Accutane); das Antikonvulsivum Phenytoin (Dilantin, Epanutin); Psychopharmaka Diazepam, Valium, Relanium, Seduxen.
Symptome Treacher-Collins-Syndrom
Die klinischen Symptome der mandibulofaszialen Dysostose und der Grad ihrer Ausprägung hängen größtenteils von den Merkmalen der Manifestation von Genmutationen ab. Die ersten Anzeichen dieser Anomalie sind in den meisten Fällen bei einem Kind unmittelbar nach der Geburt sichtbar: Das Gesicht mit Treacher-Collins-Syndrom hat ein charakteristisches Aussehen. Darüber hinaus sind morphologische Anomalien in der Regel bilateral und symmetrisch.
Die offensichtlichsten Symptome des Treacher-Collins-Syndroms sind:
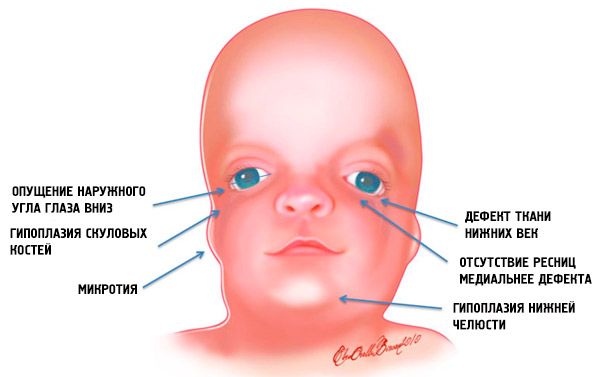
- Unterentwicklung (Hypoplasie) der Gesichtsknochen des Schädels: Jochbein, Jochfortsätze des Stirnbeins, seitliche Flügelplatten, Nasennebenhöhlen, Unterkiefer und Vorsprünge der Knochenepiphysen (Kondylen);
- Unterentwicklung der Unterkieferknochen (Mikrognathie) und ein stumpferer Unterkieferwinkel als üblich;
- die Nase hat eine normale Größe, erscheint aber aufgrund einer Hypoplasie der Augenbrauenbögen und einer Unterentwicklung oder Abwesenheit der Jochbögen in der Schläfenregion groß;
- die Augenschlitze sind nach unten gerichtet, d. h. die Form der Augen ist abnormal, wobei die äußeren Augenwinkel nach unten hängen;
- Defekte der unteren Augenlider (Kolobom) und teilweises Fehlen der Wimpern;
- unregelmäßig geformte Ohrmuscheln mit einer Vielzahl von Abweichungen, einschließlich ihrer Lage im Unterkieferwinkel, Fehlen von Ohrläppchen, blinden Fisteln zwischen dem Tragus des Ohres und dem Mundwinkel usw.;
- Verengung oder Verschluss (Atresie) des äußeren Gehörgangs und Anomalien der Gehörknöchelchen des Mittelohrs;
- Fehlen oder Hypoplasie der Ohrspeicheldrüsen;
- Rachenhypoplasie (Verengung des Rachens und der Atemwege);
- Nichtverschmelzung des harten Gaumens (Gaumenspalte) sowie Fehlen, Verkürzung oder Unbeweglichkeit des weichen Gaumens.
Solche anatomischen Anomalien führen in jedem Fall zu Komplikationen. Dazu gehören funktionelle Hörstörungen in Form von Schallleitungsschwerhörigkeit oder völliger Taubheit; Sehbehinderungen aufgrund einer Fehlbildung der Augäpfel; Gaumendefekte verursachen Schwierigkeiten beim Essen und Schlucken. Es gibt Zahnfehlstellungen (Malokklusion), die mit Kieferdefekten einhergehen und wiederum Probleme beim Kauen und Artikulieren verursachen. Pathologien des weichen Gaumens erklären die nasale Stimme.
Komplikationen und Konsequenzen
Die Folgen der Kiefer- und Gesichtsanomalien beim Treacher-Collins-Syndrom bestehen darin, dass die intellektuellen Fähigkeiten des Kindes bei der Geburt normal sind, aufgrund von Hörfehlern und anderen Störungen jedoch eine sekundäre geistige Behinderung beobachtet wird.
Darüber hinaus spüren Kinder mit solchen Defekten ihre Minderwertigkeit stark und leiden, was sich negativ auf ihr Nervensystem und ihre Psyche auswirkt.
Diagnose Treacher-Collins-Syndrom
Die postnatale Diagnose des Treacher-Collins-Syndroms basiert im Wesentlichen auf klinischen Symptomen. Eine kraniofaziale Dysostose ist bei voller Ausprägung des Syndroms leicht zu erkennen. Liegen jedoch nur minimale pathologische Symptome vor, kann die korrekte Diagnose problematisch sein.
In diesem Fall sollte besonderes Augenmerk auf die Beurteilung aller mit Anomalien verbundenen Funktionen gelegt werden, insbesondere derjenigen, die die Atmung betreffen (aufgrund des Risikos einer Schlafapnoe). Die Effektivität der Ernährung und die Hämoglobin-Sauerstoffsättigung sollten ebenfalls beurteilt und überwacht werden.
Später, am 5.-6. Tag nach der Geburt, muss das Ausmaß der Hörschädigung durch eine audiologische Untersuchung festgestellt werden, die in der Entbindungsklinik durchgeführt werden sollte.
Es wird eine Untersuchung verordnet, bei der eine instrumentelle Diagnostik mittels Durchleuchtung der kraniofazialen Dysmorphologie durchgeführt wird; Pantomographie (Panorama-Röntgenaufnahme der Knochenstrukturen des Gesichtsschädels); vollständige kraniale Computertomographie in verschiedenen Projektionen; CT oder MRT des Gehirns zur Bestimmung des Zustands des inneren Gehörgangs.
Die früheste – pränatale – Diagnose von Kiefer- und Gesichtsanomalien bei Vorliegen eines Treacher-Collins-Syndroms in der Familienanamnese ist durch eine Chorionzottenbiopsie in der 10.–11. Schwangerschaftswoche möglich (bei diesem Eingriff besteht die Gefahr einer Fehlgeburt und einer Gebärmutterinfektion).
Auch bei Familienangehörigen werden Blutuntersuchungen durchgeführt, in der 16.–17. Schwangerschaftswoche wird das Fruchtwasser untersucht (transabdominale Amniozentese), in der 18.–20. Schwangerschaftswoche wird eine Fetoskopie durchgeführt und Blut aus den fetalen Gefäßen der Plazenta entnommen.
Am häufigsten wird Ultraschall jedoch zur pränatalen Diagnose dieses Syndroms beim Fötus (in der 20. bis 24. Schwangerschaftswoche) eingesetzt.
Welche Tests werden benötigt?
Differenzialdiagnose
Dieselben Methoden werden von Spezialisten verwendet, wenn eine Differentialdiagnose erforderlich ist, um das leichte Treacher-Collins-Syndrom zu erkennen und es von anderen angeborenen Anomalien der kraniofazialen Knochen abzugrenzen, insbesondere: Apert-, Crouzon-, Nager-, Peters-Hewels-, Hellermann-Steph-Syndrom sowie Hemifaziale Mikrosomie (Goldenhar-Syndrom), Hypertelorismus, vorzeitige Fusion der Schädelnähte (Kraniosynostose) oder gestörte Fusion der Gesichtsknochen (Kraniosynostose).
Wen kann ich kontaktieren?
Behandlung Treacher-Collins-Syndrom
Wie bei allen Fällen genetisch bedingter angeborener Defekte ist die Behandlung schwerer Formen des Treacher-Collins-Syndroms ausschließlich palliativ, da es für solche Pathologien schlicht keine therapeutischen Methoden gibt. Das Spektrum und der Grad der Deformationen bei diesem Syndrom sind umfangreich, daher sind auch Art und Intensität der medizinischen Intervention vielfältig.
Zur Korrektur und Verbesserung des Hörvermögens werden Hörgeräte eingesetzt, zur Verbesserung der Sprache dienen Sprachtherapiesitzungen.
Bei schweren Verengungen der Atemwege (Tracheostomie) und des Kehlkopfes (Gastrostomie zur Ernährung) sind frühzeitig chirurgische Eingriffe erforderlich. Auch eine chirurgische Korrektur des Gaumens kann erforderlich sein.
Unterkieferverlängerungsoperationen werden im Alter von 2–3 Jahren oder später durchgeführt. Die Weichteilrekonstruktion umfasst die Korrektur eines Unterlidkoloboms und eine plastische Ohrmuscheloperation.
Verhütung
Zur Vorbeugung des Treacher-Collins-Syndroms ist es erforderlich, dass zukünftige Eltern eine genetische Beratung in Anspruch nehmen. Wenn das Syndrom in der Familie vorkommt, stellt sich die Frage nach der Möglichkeit einer Schwangerschaft, um die Geburt eines Kindes mit kraniofazialen Anomalien zu vermeiden.
Prognose
Wie ist die Prognose dieser Erkrankung? Sie hängt vom Grad der Deformation und der Intensität der Symptome ab. Das Treacher-Collins-Syndrom ist eine lebenslange Diagnose.
 [ 25 ]
[ 25 ]