Facharzt des Artikels

Neue Veröffentlichungen
Hypophysenanämie (Zwergwuchs)
Zuletzt überprüft: 12.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
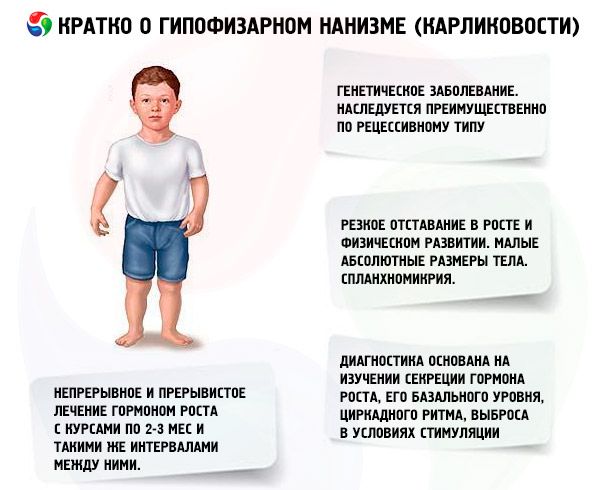
Der Begriff „Hypophysen-Zwergwuchs“ (von griechisch nanos – Zwerg; Syn.: Zwergwuchs, Nanosomie, Mikrosomie) bezeichnet im absoluten Sinne eine Erkrankung, deren Hauptmanifestation eine starke Wachstumsverzögerung ist, die mit einer Verletzung der Wachstumshormonsekretion durch den Hypophysenvorderlappen verbunden ist.
Im weiteren Sinne ist Zwergwuchs eine Wachstums- und körperliche Entwicklungsstörung, deren Auftreten nicht nur durch einen absoluten oder relativen Mangel an somatotropem Hormon aufgrund einer Pathologie der Hypophyse selbst verursacht werden kann, sondern auch durch eine Störung der hypothalamischen (zerebralen) Regulierung seiner Funktionen, Defekte in der Biosynthese des somatotropen Hormons und Störungen der Gewebeempfindlichkeit gegenüber diesem Hormon.
 [
[ Ursachen Zwergwuchs
Die meisten Formen des hypophysären Kleinwuchses sind genetisch bedingt. Am häufigsten ist der panhypopituitäre Kleinwuchs, der überwiegend rezessiv vererbt wird. Es wird angenommen, dass es zwei Übertragungswege für diese Krankheitsform gibt – autosomal und über das X-Chromosom. Bei dieser Form des Kleinwuchses ist neben dem Defekt in der Sekretion des somatotropen Hormons am häufigsten auch die Sekretion von Gonadotropinen und des Schilddrüsen-stimulierenden Hormons gestört. Die ACTH-Sekretion ist seltener und in geringerem Ausmaß gestört. Funktionelle Studien mit Releasing-Hormonen, einschließlich synthetischem Somatotropin-Releasing-Hormon (bestehend aus 29, 40 und 44 Aminosäureresten), ähnlich dem Pankreaspolypeptid, haben gezeigt, dass die meisten dieser Patienten eine Pathologie auf Hypothalamusebene aufweisen und die Insuffizienz des Hypophysenvorderlappens sekundär ist. Primäre Pathologien der Hypophyse selbst sind seltener.
Genetischer Zwergwuchs mit isoliertem Wachstumshormonmangel, beeinträchtigter biologischer Aktivität und Sensibilität dafür, tritt sporadisch in Russland und den Nachbarländern auf. Er ist häufiger auf dem amerikanischen Kontinent, in den Ländern des Nahen und Mittleren Ostens sowie in Afrika. Basierend auf den Ergebnissen einer Studie über den Blutgehalt des somatotropen Hormons und die Sensibilität der Patienten gegenüber exogenem somatotropen Hormon, den Spiegel von immunreaktivem Insulin (IRI), insulinähnlichen Wachstumsfaktoren (IGF) Typ I (Somatomedin C) und Typ II sowie die Reaktion von IGF-1 auf die Behandlung mit somatotropen Hormonpräparaten wurden verschiedene Varianten klinisch ähnlicher Zwergwuchsarten identifiziert.
Kürzlich wurde die Pathogenese des Laron-Zwergwuchses, der durch einen Mangel an IRF-1 und IRF-II verursacht wird, entschlüsselt, ebenso wie die Pathogenese des Zwergwuchses bei afrikanischen Pygmäen, der mit einem Mangel an IRF-1 und IRF-II in Zusammenhang steht.
Im Jahr 1984 wurde eine neue Variante des pseudohypophysären Zwergwuchses mit einem hohen Spiegel an somatotropem Hormon und IGF-1 beschrieben; die Entstehung des Zwergwuchses wird durch einen Defekt seiner Rezeptoren erklärt, der durch eine starke Abnahme der Bindung von Hautfibroblasten an IGF-1 nachgewiesen wird.
Es sollte betont werden, dass unter modernen Bedingungen, wo es kleine Familien gibt, viele isolierte („idiopathische“, sporadische) Fälle der Krankheit auch genetisch bedingt sein können.
Bei der Analyse von 350 Fallgeschichten war die Ätiologie des Zwergwuchses bei 228 Patienten (65,2%) unklar. Diese Gruppe umfasste Patienten aus 57 Familien mit wiederholtem Auftreten von Zwergwuchs (2-4 Fälle pro Familie), was 28 % aller Patienten ausmachte. In 77 % der Familien mit ätiologisch unklaren (meist genetischen) Formen des Zwergwuchses wurde ein unbestreitbarer Zusammenhang mit der Vererbung des Fehlens des Rh-Faktors festgestellt. Die Verteilung des Rh-Faktors in Familien von Patienten mit Zwergwuchs unterscheidet sich von der im Rh-Konflikt zwischen Mutter und Fötus beobachteten und geht in der Regel nicht mit einer hämolytischen Erkrankung des Neugeborenen einher (der Vater kann Rh-negativ sein, und im Falle der Heterozygotie der Eltern für den Rh-Faktor - die Kinder usw.). Es ist möglich, einen Zusammenhang zwischen der Aktivität von Genen, die für die Synthese des somatotropen Hormons (oder Somatotropin-Releasing-Hormons) verantwortlich sind, und Genen, die den Rh-Faktor bestimmen, anzunehmen, zumal die meisten Formen von Zwergwuchs und das Fehlen des Rh-Faktors autosomal-rezessive Merkmale sind. Dies erklärt nicht die relative Seltenheit von Zwergwuchs im Vergleich zur Häufigkeit Rh-negativer Personen in der Bevölkerung. Wahrscheinlich sind einige noch unbekannte zusätzliche Faktoren wichtig, aber die Merkmale der Verteilung des Rh-Faktors in Familien von Patienten mit familiärem und sporadischem Zwergwuchs sind wahrscheinlich nicht zufällig.
Eine große Gruppe von Patienten mit Kleinwuchs (primär zerebral, zerebral-hypophysären) sind Patienten mit verschiedenen Arten von organischen Pathologien des zentralen Nervensystems, die in der Gebärmutter oder in der frühen Kindheit entstanden. Anatomische Substrat, das diese Pathologie verursacht kann Unterentwicklung oder das Fehlen der Hypophyse, ihre Dystopie in Pathologie der Bildung der Sella turcica, zystische Degeneration der Hypophyse, ihre Atrophie durch Kompression durch einen Tumor (Kraniopharyngeom, chromophobe Adenom, Meningiom, Gliom) sein. Kleinwuchs kann durch traumatische Verletzungen der Hypothalamus-Hypophysen-Region (intrauterin, während der Geburt oder nach der Geburt) verursacht werden, die oft bei Mehrlingsschwangerschaften auftritt, sowie bei der Geburt in Steißlage, Fußlage oder in Querlage mit Rotation am Bein (dies ist der Mechanismus der Geburt bei mehr als 1/3 der Patienten mit Kleinwuchs). Wichtig sind infektiöse und toxische Schäden (intrauterine Virusinfektionen, Tuberkulose, Syphilis, Malaria, Toxoplasmose; Erkrankungen im frühen Kindesalter, Neugeborenensepsis, Meningo- und Arachnoenzephalitis usw.). Diese Prozesse können die Hypophyse selbst und die hypothalamischen Zentren, die ihre Funktion regulieren, schädigen und normale funktionelle Verbindungen im Zentralnervensystem stören.
Intrauterine fetale Läsionen können zur Geburt von Patienten mit „Zwergwuchs von Geburt an“ mit normaler Wachstumshormonsekretion führen (zerebraler primordialer Zwergwuchs, Mikrozephalie, Silver-Russell-Zwergwuchs mit Körperhemiasymmetrie und hohen Gonadotropinspiegeln usw.).
Weitere Faktoren, die die körperliche Entwicklungsstörung bei Kleinwuchs verschlimmern, können eine unzureichende Ernährung sein, die hinsichtlich essentieller Inhaltsstoffe (Proteinmangel) und Mikroelementen (Zinkmangel) unausgewogen ist, sowie ungünstige Umweltbedingungen und verschiedene chronische Erkrankungen, wie z. B. Glomerulonephritis, bei der Azotämie die Aktivität der Leberrezeptoren beeinträchtigt oder den Stoffwechsel der Leberzellen direkt beeinflusst, was zu einer Verringerung der Somatomedinsynthese führt, oder Leberzirrhose, wenn die Somatomedinbildung beeinträchtigt ist.
Pathogenese
Bei den meisten Patienten mit hypophysärem Kleinwuchs beschränken sich die Veränderungen nicht auf die Pathologie der Sekretion des somatotropen Hormons und der Empfindlichkeit gegenüber diesem, sondern erstrecken sich auf andere tropische Hormone der Hypophyse, was verschiedene Kombinationen von endokrinen und metabolischen Störungen verursacht.
Bei isoliertem Mangel an somatotropem Hormon sind morphologische Veränderungen der Hypophyse unzureichend erforscht. In den untersuchten Fällen wurden selten pathologische Störungen (Kraniopharyngeom oder Hyperostose der Schädelknochen) gefunden. Bei dieser Art von Kleinwuchs kann eine angeborene Unterentwicklung peptiderger Zellen oder ein Defekt im Neurotransmittersystem des Hypothalamus vorliegen. In solchen Fällen kann Kleinwuchs mit Dysplasie oder Hypoplasie der Sehnerven einhergehen. Intraselläre Zysten, Hypophysen- und Hypothalamustumoren führen zu einem STH-Mangel, der eine Kompression des Hypophysengewebes, insbesondere der Somatotrophen, verursacht.
Zwergwuchs ist gekennzeichnet durch eine Ausdünnung der Knochen, hauptsächlich aufgrund der Kortikalis, sowie eine verzögerte Differenzierung und Verknöcherung des Skeletts. Die inneren Organe sind hypoplastisch, manchmal atrophisch, und die Muskeln sind schlecht entwickelt.
Symptome Zwergwuchs
Eine starke Wachstumsverzögerung und körperliche Entwicklungsverzögerung sind die Hauptsymptome von hypophysärem Zwergwuchs. Patienten werden mit normalem Körpergewicht und normaler Körpergröße geboren und beginnen im Alter von 2-4 Jahren, im Wachstum zurückzubleiben.
Vor der Einführung aktiver Behandlungen gegen Nanismus galten Zwerge als Personen mit einer Körpergröße von weniger als 120 cm bei Frauen und 130 cm bei Männern. Derzeit weicht die Körpergröße eines Zwerges um mindestens 2–3 Sigma von der durchschnittlichen tabellarischen Norm für ein bestimmtes Geschlecht, Alter oder eine bestimmte Population ab. Es gibt auch eine Methode zur grafischen Bewertung der Körpergröße basierend auf der Gaußschen Verteilungskurve. In diesem Fall werden Zwerge nach Körpergröße in eine Gruppe aufgenommen, die die Mindestanzahl von Individuen in der entsprechenden Population mit der größten Abweichung von der durchschnittlichen Wachstumsnorm umfasst.
Hypophysärer Zwergwuchs ist nicht nur durch eine geringe absolute Körpergröße, sondern auch durch geringes jährliches Wachstum und eine geringe körperliche Entwicklungsdynamik gekennzeichnet. Der Körperbau ist proportional, aber die Proportionen des Körpers der Patienten sind typisch für die Kindheit. Die Haut ist blass, oft gelblich gefärbt und trocken, was auf eine absolute oder relative Schilddrüseninsuffizienz zurückzuführen ist. Manchmal wird Zyanose beobachtet - eine "Marmorierung" der Haut. Bei unbehandelten Patienten tritt frühzeitig eine altersbedingte und faltige Haut (Gerodermie) auf. Dies ist auf die unzureichende anabolische Wirkung von STH und den langsamen Wechsel der Zellgenerationen zurückzuführen.
Das Kopfhaar kann normal oder trocken, dünn und brüchig sein; lange Wimpern sind typisch. Sekundäres Haarwachstum fehlt oft. Die Größe der Sella turcica ist bei den meisten Patienten mit Zwergwuchs (70-75%) unverändert, aber die Sella behält oft die kindliche Form eines „stehenden Ovals“, hat einen breiten „jugendlichen“ Rücken, der Keilbeinhöhlensinus hinkt hinsichtlich der Pneumatisierung hinterher. Es gibt jedoch Patienten mit einer vergrößerten Sella turcica, was ein Zeichen für einen Tumor ist; mit Verkalkungsbereichen im Hintergrund oder im Eingangsbereich (bei Kraniopharyngeom, Resteffekten einer Neuroinfektion) oder deren Verkleinerung (Anzeichen einer Unterentwicklung, geringe Größe der Hypophyse). Symptome einer intrakraniellen Hypertonie werden beobachtet: Ausdünnung der Schädelknochen, verstärktes Gefäßmuster, Vorhandensein von Fingerabdrücken usw. Das wichtigste Anzeichen für hypophysären Zwergwuchs ist eine Verzögerung der Differenzierung und Verknöcherung des Skeletts. Merkmale des Zahnsystems hängen ebenfalls eng mit der Skelettdifferenzierung zusammen: Es wird ein später Ersatz der Milchzähne festgestellt. Die größte Verzögerung in der Entwicklung des Skelettsystems wird bei Patienten mit Zwergwuchs mit sexueller Insuffizienz und Hypothyreose beobachtet.
Die Genitalien der meisten Patienten sind stark unterentwickelt, Fehlbildungen sind jedoch selten. Wir beobachteten Kryptorchismus bei 5,8 % der männlichen Patienten. Sexuelle Insuffizienz geht mit einer Unterentwicklung der sekundären Geschlechtsmerkmale, vermindertem Sexualverlangen und Ausbleiben der Menstruation einher. Eine normale spontane sexuelle Entwicklung wird nur bei Patienten mit isoliertem Wachstumshormonmangel und bei einigen Patienten mit zerebralem Kleinwuchs beobachtet.
Schilddrüseninsuffizienz ist ein recht häufiges Symptom für Zwergwuchs. Es ist zu beachten, dass die äußeren Erscheinungsformen einer Hypothyreose nicht immer den wahren Funktionszustand der Schilddrüse widerspiegeln. Dies ist auf eine relative Hypothyreose zurückzuführen, die auf eine Verletzung des Übergangs von Thyroxin (T 4 ) zu Trijodthyronin (T 3 ) und die Bildung von inaktivem (reversiblem) T 3 zurückzuführen ist, was für eine somatotrope Insuffizienz charakteristisch ist.
Die adrenocorticotrope Funktion nimmt bei hypophysärem Kleinwuchs seltener und in geringerem Ausmaß ab als die sexual- und schilddrüsenstimulierenden Funktionen und erfordert bei den meisten Patienten keine besondere Korrektur.
In den meisten Fällen ist der Intellekt nicht beeinträchtigt. Es treten emotionale Veränderungen in Form von mentalem Infantilismus auf; bei älteren Patienten ohne intellektuelle Beeinträchtigung werden manchmal reaktive Neurosen beobachtet.
Bei organischer Hirnerkrankung, insbesondere tumoröser Natur, kann Kleinwuchs mit Symptomen von Diabetes insipidus, bitemporaler Hemianopsie und geistiger Behinderung auftreten.
Die Untersuchung der Entwicklung der bioelektrischen Aktivität des Gehirns bei Patienten ohne organische Symptome des Zentralnervensystems zeigte, dass ihr EEG durch Merkmale der Unreife und der langfristigen Erhaltung einer hohen "kindlichen" EEG-Spannung gekennzeichnet ist. Ungleichmäßigkeit des Alpha-Rhythmus in Amplitude und Frequenz; ein starker Anstieg des Gehalts an langsamen θ- und δ-Rhythmen, insbesondere in den frontalen und zentralen Ableitungen; eine deutliche Reaktion auf Hyperventilation; eine Verschiebung des Bereichs der EEG-Rhythmen nach den Rhythmen der Lichtstimulation hin zu niedrigen Frequenzen (Hinweis auf eine Abnahme der funktionellen Beweglichkeit der Nervenstrukturen des Gehirns). Es zeigte sich, dass bei älteren Patienten die Unreife der elektrischen Aktivität des Gehirns auf eine sexuelle Unterentwicklung und bei Patienten aller Altersgruppen auf eine Hypothyreose zurückzuführen ist.
Der Kohlenhydratstoffwechsel bei Patienten mit Zwergwuchs ist gekennzeichnet durch eine Tendenz zur Senkung des Nüchternblutzuckerspiegels, einen Anstieg bei körperlicher Anstrengung, einen Mangel an endogenem Insulin, eine erhöhte Empfindlichkeit gegenüber exogenem Insulin mit häufiger Entwicklung von hypoglykämischen Zuständen. Letzteres ist hauptsächlich auf einen unzureichenden Spiegel an gegeninsulären Hormonen im Körper der Patienten zurückzuführen.
Die inneren Organe zeigen Splanchnomykrie, d. h. eine Verkleinerung. Es wurden keine für Kleinwuchs spezifischen funktionellen Veränderungen der inneren Organe beschrieben. Häufig tritt eine arterielle Hypotonie mit vermindertem systolischen und diastolischen Druck und verringerter Pulsamplitude auf. Die Herztöne sind gedämpft, aufgrund trophischer Veränderungen im Myokard und autonomer Störungen sind funktionelle Geräusche verschiedener Art zu hören. Das EKG ist gekennzeichnet durch Niederspannung (insbesondere bei Hypothyreose), Sinusbradykardie oder Bradyarrhythmie; das PCG zeigt eine Abnahme der Amplitude von Tönen, zusätzlichen Tönen und funktionellen Geräuschen. Daten der Oxygemometrie weisen auf Hypoxämie (anfänglich und während körperlicher Anstrengung) und Sauerstoffmangel hin. Ältere Patienten entwickeln manchmal Bluthochdruck.
Diagnose Zwergwuchs
Die Diagnose und Differentialdiagnose von Zwergwuchs basiert auf Anamnesedaten und einer umfassenden klinischen, radiologischen, labortechnischen und hormonellen Untersuchung. Neben der absoluten Körpergröße wird zur Beurteilung des Wachstums der Patienten auch das Wachstumsdefizit ermittelt - die Differenz zwischen der Körpergröße des Patienten und seiner durchschnittlichen Norm für das entsprechende Geschlecht und Alter; Wachstumsalter - Übereinstimmung der Körpergröße des Patienten mit bestimmten Standards; der Indikator der normalisierten Abweichung
I = M - Mcp / δ, wobei M die Körpergröße des Patienten, Mcp die durchschnittliche Normalgröße für ein bestimmtes Geschlecht und Alter und δ die quadratische Abweichung von Mcp ist; I kleiner als 3 ist typisch für Nanismus, I größer als 3 ist typisch für Gigantismus. Dieser Indikator kann zur Beurteilung der Entwicklungsdynamik verwendet werden.
Röntgenuntersuchungen von Patienten mit Kleinwuchs zeigen Anzeichen von intrakranieller Hypertonie, Restfolgen einer Neuroinfektion, Verkalkungen und Kraniosynostose. Die Untersuchung von Größe, Form und Struktur der Sella turcica gilt als indirekter Indikator zur Charakterisierung der Hypophysengröße. Eine der wichtigsten Manifestationen einer pathologischen Wachstumsverzögerung ist eine Verletzung der Skelettdifferenzierung. Zur Beurteilung des Skelettreifegrades wird das (röntgenologische) Knochenalter bestimmt, das der Differenzierung des Knochengewebes entspricht; der Ossifikationsmangel ist der Grad der Ossifikationsverzögerung von der Norm (in Jahren), der Ossifikationskoeffizient ist der Quotient aus Knochenalter dividiert durch chronologische und andere Parameter.
Die moderne Diagnostik von Kleinwuchs ist ohne die Untersuchung der Sekretion des somatotropen Hormons, seines Basalspiegels, seines zirkadianen Rhythmus und seiner Freisetzung unter Stimulation nicht möglich. Die meisten Patienten mit hypophysärem Kleinwuchs weisen einen verringerten Gehalt an somatotropem Hormon im Blutserum auf. Bei radioimmunologischer Bestimmung liegt er (verschiedenen Autoren zufolge) zwischen (0,87 ± 0,09) und (1,50 ± 0,64) ng/ml, mit einem durchschnittlichen Normwert von (3,81 ± 0,29) ng/ml. Eine Untersuchung der täglichen (zirkadianen) Rhythmen der Sekretion des somatotropen Hormons hat gezeigt, dass dessen Spiegel bei gesunden Menschen während der ersten beiden Schlafstunden sowie zwischen 4 und 6 Uhr morgens am höchsten ist. Bei Kleinwuchs ist der Gehalt an somatotropem Hormon auch während dieser Stunden verringert.
Zur Untersuchung der Reserven der somatotropen Funktion werden verschiedene Stimulanzien verwendet und der Gehalt an somatotropem Hormon vor und nach ihrer Verabreichung untersucht. Blut für die Untersuchung wird 2-3 Stunden lang alle 30 Minuten entnommen. Die Freisetzung von somatotropem Hormon nach der Stimulation gilt als normal, mindestens bis zu 7-10 ng/ml, manchmal erreicht sie 20-40 ng/ml. Tritt in einer der Proben keine Reaktion auf, werden Wiederholungstests mit anderen Stimulanzien durchgeführt. Ein Mangel an somatotropem Hormon gilt als nachgewiesen, wenn in 2-3 verschiedenen Proben keine Freisetzung von somatotropem Hormon festgestellt wird.
Die am häufigsten verwendeten stimulierenden Tests sind: Bei intravenöser Verabreichung von 0,1 U (0,75–1,5 U) Insulin pro 1 kg Körpergewicht des Patienten und Erreichen einer Hypoglykämie (Abnahme des Blutzuckerspiegels um 50 % gegenüber dem Ausgangswert) wird das somatotrope Hormon im Serum nach obigem Schema bestimmt. Bei schwerer Hypoglykämie wird der Test abgebrochen und dem Patienten intravenös Glukose verabreicht. Dies ist die gebräuchlichste, klassische Diagnosemethode.
TRH in einer Dosis von 200–500 µg intravenös. Identifiziert effektiv Hormonreserven, verursacht keine Komplikationen. In Kombination mit dem Insulintest ermöglicht es die Beurteilung des Schädigungsgrades des Hypothalamus-Hypophysen-Systems. Eine positive Reaktion auf TRH bei fehlender Insulin-Hypoglykämie weist auf eine intakte Hypophyse und eine Schädigung des Hypothalamus hin, negative Reaktionen auf TRH und Hypoglykämie deuten auf eine Schädigung der Hypophyse selbst hin.
TRH, LH-RH in einer intravenösen Dosis von 300 µg ist ähnlich wie die vorherige.
Humanes SGH ist ein synthetisches Analogon einer biologisch aktiven Verbindung, die aus einem Pankreastumor isoliert wurde. Derzeit gibt es 3 Arten von synthetischem SGH: mit 29, 40 und 44 Aminosäureresten. Es wird intravenös in Dosen von 1 bis 3 µg/kg Körpergewicht des Patienten angewendet. Die Freisetzung von STH wird 15-20 Minuten nach der Verabreichung beobachtet, der Test ist wirksamer als andere beim Nachweis von Reserven an endogenem Wachstumshormon. Eine positive SGH-Reaktion weist auf eine hypothalamische Schädigung der somatotropen Funktion und eine intakte Hypophyse hin; mit Aminosäuren (L-Argininmonochlorid, Ornithin, Tryptophan, Glycin, Leucin) intravenös in einer Dosis von 0,25-0,5 g pro 1 kg Körpergewicht des Patienten. Wirksam zur Untersuchung von SGH-Reserven. Kann allergische Reaktionen hervorrufen.
L-Dopa oral in einer Dosis von 250-500 µg. Wirksam, von den Patienten gut vertragen.
Darüber hinaus kommen Tests mit Glucagon, Bromergocryptin (Parlodel), Lysin-Vasopressin, Clonidin und dosierter fahrradergometrischer Belastung zum Einsatz.
Die Untersuchung des Zustands der somatotropen Funktion ist nicht nur für die Diagnose von Kleinwuchs notwendig, sondern auch für die begründete Wahl einer Therapiemethode, da eine Behandlung mit Somatotropin nur bei einem Mangel an endogenem Wachstumshormon sinnvoll ist.
Für die Diagnose der Form des Zwergwuchses ist es sehr wichtig, den Gehalt an insulinähnlichen Wachstumsfaktoren oder Somatomedinen (insbesondere IGF-1 oder Somatomedin C) zu untersuchen - Mediatoren der Wirkung des somatotropen Hormons auf Gewebeebene. Es ist bekannt, dass der Gehalt an Somatomedin C bei Zwergwuchs reduziert und bei Akromegalie im Vergleich zur Norm erhöht ist. Die von Laron beschriebene Form des Zwergwuchses ist eine Art von Krankheit mit normaler Produktion von STH, aber mit einer Verletzung der Bildung von IGF-1 und IGF-II. Die Behandlung solcher Patienten mit Somatotropin ist zwecklos.
Indirekte Indikatoren für die somatotrope Funktion der Hypophyse sind die Aktivität der alkalischen Phosphatase und der Gehalt an anorganischem Phosphor im Serum. Unter hyposomatotropen Bedingungen sind diese Indikatoren reduziert. Bei der panhypopituitären Form des Zwergwuchses ist die Sekretion von Gonadotropinen, häufig TSH, verringert, was mit einer entsprechenden Abnahme der Funktionen der Geschlechtsdrüsen (Mangel an Androgenen oder Östrogenen), der Schilddrüse (Abnahme der Spiegel von T3 , T4 , proteingebundenem Jod - PBI, Ansammlung von 131 I durch die Schilddrüse) und der Nebennieren (Abnahme der Menge an Cortisol und 17-OCS im Plasma, Ausscheidung von 17-KC und 17-OCS im Urin, Lymphozytose) einhergeht.
Alle Formen des hypophysären (hypothalamisch-hypophysären) genetisch bedingten Zwergwuchses sind gekennzeichnet durch wiederholte Erkrankungen von Kindern in Familien mit autosomal-rezessiver (häufiger) oder autosomal-dominanter Vererbung, Wachstumsverzögerung und körperlicher Entwicklung im Alter von 2–4 Jahren mit einer Verzögerung von mindestens 2–3 ° gegenüber den durchschnittlichen Wachstumsnormen für ein bestimmtes Geschlecht, Alter und eine bestimmte Bevölkerungsgruppe, mit geringer spontaner jährlicher Wachstumsdynamik und verzögerter Ossifikation. Bei einem niedrigen somatotropen Hormonspiegel (in 2–3 Stimulationstests unter 7 ng/ml) ist die Therapie mit somatotropem Hormon hochwirksam (ergibt eine Körpergrößenzunahme von mindestens 7 cm pro Jahr). Bei einem normalen oder hohen somatotropen Hormonspiegel (mit seiner biologischen Inaktivität) kann die Empfindlichkeit gegenüber dem Hormon erhalten bleiben. Es werden keine Veränderungen der Intelligenz beobachtet.
Bei genetisch bedingtem Zwergwuchs mit Gewebeunempfindlichkeit gegenüber somatotropem Hormon ähnelt das klinische Bild einem isolierten Wachstumshormonmangel, eine Somatotropintherapie ist jedoch wirkungslos. In dieser Gruppe lassen sich je nach IRF-Spiegel folgende Hauptformen unterscheiden: mit normalem Gehalt (IRF-Rezeptordefekt) und reduziertem – Zwergwuchs vom Laron-Typ (IRF-1- und IRF-II-Mangel) und der bei afrikanischen Pygmäen vorkommende Typ (IRF-1-Mangel).
Zerebraler Zwergwuchs ist durch isolierte Erkrankungen in einer Familie gekennzeichnet, die mit intrauterinen oder postnatalen Schäden des Zentralnervensystems verbunden sind, wobei offensichtliche organische Veränderungen im Zentralnervensystem vorliegen, oft kombiniert mit einer Pathologie des Sehorgans, dem Vorliegen von Diabetes insipidus, der Erhaltung der gonadotropen Funktionen und Veränderungen der Intelligenz.
Einige Formen der Gonadendysgenesie und -agenesie gehen mit ausgeprägter Kleinwüchsigkeit einher, insbesondere das Shereshevsky-Turner-Syndrom und die „Turneroid“-Form (Mosaikform) der Hodendysgenesie. Zytogenetische Untersuchungen (Geschlechtschromatin, Karyotyp) helfen bei der Differentialdiagnose und decken Chromosomendefekte sowie charakteristische Defekte der somatischen und sexuellen Entwicklung, normale oder erhöhte Spiegel des endogenen somatotropen Hormons und Unempfindlichkeit gegenüber der Behandlung mit Somatotropin auf.
Unter den endokrinen Störungen, die bei Kleinwuchs auftreten, ist die primäre Hypothyreose hervorzuheben, die durch angeborene Hypoplasie oder Aplasie der Schilddrüse, deren Dystopie, enzymatische Defekte in der Biosynthese von Schilddrüsenhormonen und frühe Autoimmunschäden an der Schilddrüse verursacht wird. Bei all diesen Erkrankungen dominieren Anzeichen einer Hypothyreose mit hohem TSH-Spiegel, einer Abnahme von T4 und T3 im Blutserum . Bei einem Myxödem autoimmuner Genese werden Antikörper gegen Thyreoglobulin, mikrosomale und nukleare Fraktionen des Schilddrüsengewebes im Blut nachgewiesen, der Spiegel des somatotropen Hormons ist normal oder erniedrigt. Der klinische Effekt kann durch die ausschließliche Kompensation der Hypothyreose erzielt werden.
Kleinwuchs geht mit vorzeitiger sexueller Entwicklung und Adrenogenitalsyndrom aufgrund der frühen Schließung von Wachstumszonen einher; Itsenko-Cushing-Krankheit, die im Kindesalter aufgrund der hemmenden Wirkung von Glukokortikoiden auf die Sekretion des somatotropen Hormons und ihrer katabolen Wirkung auftritt; Mauriac-Syndrom - Kleinwuchs und Infantilismus bei Patienten mit schwerem insulinabhängigem Diabetes mellitus.
Hypophysen-Zwergwuchs sollte von somatogener Verzögerung der körperlichen Entwicklung unterschieden werden, die durch chronische Stoffwechselstörungen (bei Erkrankungen der Leber, der Nieren, des Magen-Darm-Trakts), chronische Hypoxie (bei Erkrankungen des Herz-Kreislauf- und Atmungssystems, bei Anämie) verursacht wird; mit systemischen Erkrankungen des Bewegungsapparates (Chondrodystrophie, unvollständige Osteogenese, Exostose-Krankheit) usw.
Eine funktionelle (konstitutionelle) Wachstumsverzögerung wird manchmal mit spätem Beginn der Pubertät bei scheinbar gesunden Jugendlichen beobachtet; wir haben festgestellt, dass sie primär mit einer vorübergehenden Insuffizienz der gonadotropen Aktivität zusammenhängt. Die Sekretion des somatotropen Hormons ist in der Regel nicht beeinträchtigt oder leicht reduziert. Die Stimulation von Gonadotropinen kann sowohl die sexuelle Entwicklung als auch das Wachstum beschleunigen.
Kleinwuchs familiärer Natur sollte als Variante der physiologischen Entwicklung betrachtet werden.
Was muss untersucht werden?
Wie zu prüfen?
Welche Tests werden benötigt?
Wen kann ich kontaktieren?
Behandlung Zwergwuchs
Die Behandlung von Kleinwuchs ist ein langwieriger Prozess. Dies zwingt den Arzt, die Mittel zur Beeinflussung des Wachstums über einen längeren Zeitraum zu verteilen, um den größtmöglichen klinischen Effekt zu erzielen und dabei zwei Grundprinzipien zu beachten:
- maximale Annäherung der behandlungsbedingten Entwicklung an physiologische Verhältnisse;
- unter Schonung der Epiphysenwachstumszonen.
Aufgrund unserer langjährigen Erfahrung in der Behandlung von Kleinwuchs halten wir das folgende Schema einer mehrstufigen Therapie für angemessen. Die Diagnose von Kleinwuchs bei erwachsenen Patienten ist in der Regel unzweifelhaft. Bei Kleinkindern ist bei unklarem Krankheitsbild eine 6- bis 12-monatige Beobachtungsphase ohne Hormontherapie erforderlich. Während dieser Zeit wird eine umfassende, allgemein kräftigende Behandlung verordnet; eine ausreichende Ernährung mit erhöhtem Gehalt an tierischem Eiweiß, Obst und Gemüse, Vitamin A und D sowie Kalzium- und Phosphorpräparaten ist erforderlich. Das Fehlen ausreichender Veränderungen des Wachstums und der körperlichen Entwicklung vor diesem Hintergrund sowie das Erkennen endokriner Störungen bei der Untersuchung bilden die Grundlage für den Beginn einer Hormontherapie.
Die wichtigste pathogenetische Therapieform bei hypophysärem Zwergwuchs ist die Gabe von menschlichem Wachstumshormon, da die meisten Fälle von Zwergwuchs zweifellos von der einen oder anderen Form eines Mangels abhängen. Aufgrund der Speziesspezifität dieses Hormons ist nur Somatotropin von Menschen und Primaten beim Menschen wirksam. In der Klinik wird häufig ein Medikament verwendet, das aus der Hypophyse von Patienten isoliert wird, die an nichtinfektiösen und nichtneoplastischen Erkrankungen gestorben sind. Menschliches Somatotropin wird durch bakterielle Synthese unter Verwendung von Escherichia coli mittels Gentechnik gewonnen. Menschliches Somatotropin wird auch chemisch synthetisiert, ist aber extrem teuer und wird in der Klinik praktisch nicht verwendet. Für die Somatotropin-Behandlung werden Patienten mit nachgewiesenem Mangel an endogenem Wachstumshormon und einer Skelettdifferenzierung ausgewählt, die das für 13- bis 14-Jährige typische Niveau nicht überschreitet. Für die Behandlung gibt es keine Altersbeschränkung.
Die minimalen wirksamen Dosen, die in der ersten Behandlungsphase angewendet werden können, betragen 0,03–0,06 mg/kg Körpergewicht. Die wirksamsten Dosen sind 2–4 mg dreimal pro Woche. Eine Erhöhung der Einzeldosis auf 10 mg führte nicht zu einer ausreichenden Steigerung des Wachstumseffekts, verursachte jedoch eine schnelle Bildung von Antikörpern gegen Somatotropin.
In unserem Land wird seit 1960 an der Erforschung des menschlichen Wachstumshormons gearbeitet. Es wurden zwei Behandlungsschemata getestet: kontinuierlich und intermittierend mit Kuren von 2–3 Monaten und gleichen Abständen dazwischen. Die durchschnittliche Größenzunahme der Patienten während des 1. Behandlungsjahres betrug 9,52 ± 0,39 cm, die Zunahme des Körpergewichts betrug 4,4 ± 0,14 kg. Bei langfristiger kontinuierlicher Behandlung betrug die durchschnittliche Größenzunahme 0,82 cm/Monat, das Körpergewicht 0,38 kg/Monat; bei intermittierender Behandlung 0,75 cm/Monat bzw. 0,4 kg/Monat. Die kontinuierliche Behandlung führte zu einem schnelleren Größenzuwachs mit einem starken Rückgang der Wirkung nach 1–1,5 Jahren, bei intermittierender Behandlung blieb die Wirksamkeit 3–4 Jahre lang erhalten, sodass wir das Kurbehandlungsschema als geeigneter erachten. Die Bestimmung des IGF-I-Spiegels (Somatomedin C) kann als zuverlässiger Indikator für die Empfindlichkeit des Patienten gegenüber der Behandlung mit Somatotropin-Medikamenten dienen. Ein Anstieg des IGF-I-Gehalts nach der Einführung des somatotropen Hormons lässt einen positiven Therapieeffekt erwarten. Ein wichtiger Vorteil der Somatotropin-Behandlung ist das Fehlen einer Beschleunigung der Skelettverknöcherung vor diesem Hintergrund.
Das wichtigste Mittel zur Behandlung von Zwergwuchs ist die Verwendung von anabolen Steroiden, die das Wachstum stimulieren, indem sie die Proteinsynthese verbessern und den Spiegel des endogenen somatotropen Hormons erhöhen. Die Behandlung wird über mehrere Jahre durchgeführt, wobei einige Medikamente schrittweise durch andere ersetzt werden, von weniger aktiven zu aktiveren Verbindungen. Ein Wechsel der anabolen Medikamente ist angezeigt, wenn der Wachstumseffekt nach 2-3 Jahren nachlässt, was zu einer zusätzlichen Wachstumssteigerung führt. Die Behandlung wird in Kursen durchgeführt (die Ruhezeit sollte die Hälfte der Behandlungszeit betragen). Im Falle einer Abhängigkeit sind auch längere Pausen angezeigt (bis zu 4-6 Monate). Es wird jeweils nur eines der anabolen Steroide verschrieben. Die Kombination von 2 oder mehr Medikamenten ist nicht ratsam, da dies ihre Stoffwechsel- und Wachstumseffekte nicht verbessert. Letzteres hängt in erster Linie vom Alter der Patienten und dem Differenzierungsgrad der Skelettknochen zu Beginn der Behandlung ab. Die beste Wirkung wird bei Patienten unter 16–18 Jahren beobachtet, deren Skelettverknöcherung das für 14-Jährige charakteristische Niveau nicht überschreitet. Es wird empfohlen, die Behandlung unmittelbar nach der Diagnose zu beginnen, normalerweise im Alter von 5–7 Jahren. Vor der Behandlung muss die Verschreibung von Gonadotropinen und Sexualhormonen vermieden werden, da diese das Wachstum stimulieren und gleichzeitig die Skelettdifferenzierung beschleunigen. Das Dosierungsprinzip anaboler Steroide besteht darin, von der minimal wirksamen Dosis zu schrittweise ansteigenden Dosen zu gelangen. Die empfohlenen Dosen der gängigsten Medikamente: Nerobol (Methandrostenol, Dianabol) – 0,1–0,15 mg pro 1 kg Körpergewicht pro Tag oral; Nerobolil (Durabolin) – 1 mg pro 1 kg Körpergewicht pro Monat intramuskulär, die monatliche Dosis wird in 2–3 Dosen nach 15 bzw. 10 Tagen verabreicht; Retabolil (Deca-Durabolin) – 1 mg pro 1 kg Körpergewicht einmal monatlich intramuskulär. Eine Überschreitung der angegebenen Dosen kann zur Androgenisierung führen. In physiologischen Dosen beeinflussen diese Verbindungen den Zustand der Genitalien und die Differenzierung der Skelettknochen nicht signifikant, sodass sie bei Patienten beiderlei Geschlechts über einen langen Zeitraum angewendet werden können. Mädchen sollten unter gynäkologischer Aufsicht stehen, da bei einigen Patienten im Falle einer Überdosierung oder erhöhter individueller Empfindlichkeit Anzeichen einer Virilisierung auftreten können, die sich nach Absetzen der Behandlung schnell zurückbilden. Orale Arzneimittel, die an der 17. Position methyliert bis ethyliert sind, können manchmal eine cholestatische Wirkung haben. Daher sollten bei Lebererkrankungen parenterale Anabolika bevorzugt oder orale Arzneimittel mit Choleretika kombiniert werden. Sehr selten kann die Behandlung mit Anabolika allergische Reaktionen (Juckreiz, Hautausschläge) hervorrufen. Wenn keine Komplikationen auftreten, werden Anabolika so lange angewendet, wie der Wachstumseffekt beobachtet wird (bis zu 16–18 Jahre, manchmal auch länger). Die Behandlung erfolgt vor dem Hintergrund einer allgemeinen Kräftigungstherapie.
Liegen bei den Patienten Anzeichen einer Schilddrüsenunterfunktion vor, werden ihnen gleichzeitig Schilddrüsenmedikamente (Thyroxin, Thyreoidin, Thyrotom) in individuell abgestimmter Dosierung verschrieben.
Bei der Behandlung von Jungen ist der nächste Schritt die Verabreichung von humanem Choriongonadotropin. Dieses Medikament wird frühestens im Alter von 15–16 Jahren und oft auch später angewendet, um die Leydig-Zellen zu stimulieren, was sowohl die sexuelle Entwicklung als auch das Wachstum beschleunigt (aufgrund der anabolen Wirkung ihrer eigenen Androgene). Dosen von 1000 bis 1500 IE werden 1–2 Mal pro Woche intramuskulär in 2-monatigen Kursen höchstens 2–3 Mal pro Jahr verabreicht. Wenn die Wirkung nicht vollständig ist, wird die Behandlung mit humanem Choriongonadotropin bei Jungen ab 16 Jahren mit der Verabreichung kleiner Dosen von Androgenen (Methyltestosteron in einer Dosis von 5–10 mg/Tag sublingual) abgewechselt.
Mädchen über 16 Jahre können die Behandlung mit kleinen Östrogendosen beginnen, um einen normalen Sexualzyklus zu simulieren. Die Behandlung dauert drei Wochen im Monat, gefolgt von einer Pause. In der zweiten Phase des Zyklus, ab der dritten Woche, kann Choriongonadotropin in einer Dosis von 1000-1500 IE 3-5 mal pro Woche oder Medikamente mit gestagener Wirkung (Pregnin, Progesteron) verschrieben werden.
Die letzte Phase der Behandlung (nach dem Schließen der Wachstumszonen) ist die konstante Verabreichung therapeutischer Dosen von Sexualhormonen, die dem Geschlecht des Patienten entsprechen, um die Genitalien und sekundären Geschlechtsmerkmale vollständig zu entwickeln und Libido und sexuelle Potenz sicherzustellen. Kombinierte Östrogen-Gestagen-Präparate (Non-Ovlon, Bisecurin, Infekundin, Rigevidon) eignen sich zur Behandlung weiblicher Patienten, und Androgenpräparate mit verlängerter Freisetzung (Testenat, Sustanon-250, Omnadren-250) eignen sich zur Behandlung männlicher Patienten.
Es wird eine allgemeine Kräftigungsbehandlung durchgeführt (Regime, Protein-Gemüse-Diät, Vitamintherapie, Biostimulanzien). Die Verwendung von Zinkpräparaten ist angezeigt, in deren Wirkmechanismus die Erhöhung der Aktivität von IGF-1 (insulinähnlicher Wachstumsfaktor I) die Hauptrolle spielt.
Bei organischen Erkrankungen des Zentralnervensystems werden entzündungshemmende, resorptive und Dehydratationstherapien durchgeführt. Eine gezielte systematische Therapie wirkt ermutigend. Als Ergebnis einer langfristigen, stufenweisen Behandlung erreichten 148 (80,4 %) von 175 Patienten mit Kleinwuchs beiderlei Geschlechts eine Körpergröße von über 130 cm, 92 (52,5 %) über 140 cm und 32 (18,3 %) 150–160 cm oder mehr. Gleichzeitig steigerten 37 Patienten (21,2 %) ihre Körpergröße um 30 cm, 107 (61,1 %) um 31–50 cm und 31 (17,7 %) um 51–60 cm oder mehr.
Prognose
Die Prognose hängt von der Form des Zwergwuchses ab. Bei genetisch bedingten Zwergwuchsarten ist die Lebensprognose günstig. Bei Vorliegen eines Hypophysentumors und einer organischen Schädigung des Zentralnervensystems wird dies durch die Dynamik der Entwicklung des hauptsächlichen pathologischen Prozesses bestimmt. Moderne Therapiemethoden haben die körperlichen Fähigkeiten und die Arbeitsfähigkeit der Patienten deutlich gesteigert und ihre Lebenserwartung verlängert. Während der aktiven Behandlung müssen die Patienten alle 2-3 Monate von einem Arzt untersucht werden, bei Erhaltungstherapie alle 6-12 Monate.
Für die soziale Anpassung der Patienten ist eine Beschäftigung, die ihren geistigen und körperlichen Fähigkeiten entspricht, von größter Bedeutung.
Es empfiehlt sich, Berufe zu wählen, die nicht mit schwerer körperlicher Anstrengung verbunden sind, in denen Sie aber intellektuelle Fähigkeiten, die Fähigkeit zu präziser Arbeit und Sprachkenntnisse unter Beweis stellen können.