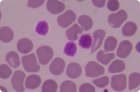
Das Von-Willebrand-Syndrom ist ein angeborener Mangel an Von-Willebrand-Faktor (VWF), der zu einer Thrombozytenfunktionsstörung führt. Es ist in der Regel durch leichte Blutungen gekennzeichnet. Screenings zeigen eine verlängerte Blutungszeit, normale Thrombozytenzahlen und möglicherweise eine leicht erhöhte partielle Thromboplastinzeit.
Thrombotisch-thrombozytopenische Purpura und hämolytisch-urämisches Syndrom sind akute, fulminante Erkrankungen, die durch die Entwicklung von Thrombozytopenie und mikroangiopathischer hämolytischer Anämie gekennzeichnet sind.
Die idiopathische (immune) thrombozytopenische Purpura ist eine hämorrhagische Erkrankung, die durch eine Thrombozytopenie verursacht wird und nicht mit einer systemischen Erkrankung assoziiert ist. Bei Erwachsenen verläuft sie meist chronisch, bei Kindern jedoch oft akut und vorübergehend. Die Milz ist normal groß.
Erbliche intrazelluläre Thrombozytenerkrankungen sind selten und führen zu lebenslangen Blutungen. Die Diagnose wird durch eine Thrombozytenaggregationsanalyse bestätigt. Bei schweren Blutungen sind Thrombozytentransfusionen notwendig.
Thrombozyten sind Fragmente von Megakaryozyten, die für die Hämostase des zirkulierenden Blutes sorgen. Thrombopoietin wird von der Leber als Reaktion auf eine Abnahme der Anzahl von Megakaryozyten im Knochenmark und zirkulierenden Thrombozyten synthetisiert und stimuliert das Knochenmark zur Synthese von Thrombozyten aus Megakaryozyten.
Zu den Folgen einer Lymphopenie gehören die Entwicklung opportunistischer Infektionen und ein erhöhtes Risiko für Krebs und Autoimmunerkrankungen. Wird im Rahmen eines Blutbildes eine Lymphopenie festgestellt, sind diagnostische Untersuchungen auf Immundefizienz und eine Analyse der Lymphozytensubpopulation erforderlich. Die Behandlung richtet sich nach der Grunderkrankung.
Neutropenie (Agranulozytose, Granulozytopenie) ist eine Abnahme der Anzahl neutrophiler Granulozyten im Blut. Bei schwerer Neutropenie steigt das Risiko und der Schweregrad bakterieller und Pilzinfektionen.
Aufgrund der hohen Inzidenz von HbS-Thalassämie und Beta-Thalassämie in bestimmten Bevölkerungsgruppen ist das angeborene Vorhandensein beider Anomalien recht häufig.
Symptome und Beschwerden sind auf die Entwicklung von Anämie, Hämolyse, Splenomegalie, Knochenmarkhyperplasie und, bei Mehrfachtransfusionen, einer Eisenüberladung zurückzuführen. Die Diagnose basiert auf einer quantitativen Hämoglobinanalyse.