Facharzt des Artikels

Neue Veröffentlichungen
Prionen - Erreger von Prionenkrankheiten
Zuletzt überprüft: 06.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Langsame Virusinfektionen zeichnen sich durch besondere Kriterien aus:
- eine ungewöhnlich lange Inkubationszeit (Monate, Jahre);
- eine spezifische Schädigung von Organen und Geweben, vor allem des zentralen Nervensystems;
- langsames, stetiges Fortschreiten der Krankheit;
- unvermeidlich tödlichen Ausgang.

Einige Erreger akuter Virusinfektionen können auch langsam verlaufende Virusinfektionen auslösen. So verursacht das Masernvirus manchmal SSPE und das Rötelnvirus progressive kongenitale Röteln und Röteln-Panenzephalitis.
Eine typische langsame Virusinfektion bei Tieren wird durch das Visna/Madi-Virus verursacht, ein Retrovirus. Es ist der Erreger einer langsamen Virusinfektion und einer fortschreitenden Lungenentzündung bei Schafen. Die weiße Substanz des Gehirns wird zerstört, es kommt zu Lähmungen (Visna – Abmagerung); es kommt zu chronischen Entzündungen der Lunge und der Milz.
Krankheiten, die in ihren Merkmalen langsamen Virusinfektionen ähneln, werden durch Prionen verursacht – die Erreger von Prioneninfektionen. Prionenkrankheiten sind eine Gruppe fortschreitender Erkrankungen des zentralen Nervensystems von Mensch und Tier. Beim Menschen ist die Funktion des zentralen Nervensystems beeinträchtigt, es treten Persönlichkeitsveränderungen und Bewegungsstörungen auf. Die Krankheitssymptome dauern in der Regel mehrere Monate bis mehrere Jahre und enden mit dem Tod. Bisher wurden Prioneninfektionen zusammen mit den sogenannten Erregern langsamer Virusinfektionen betrachtet.
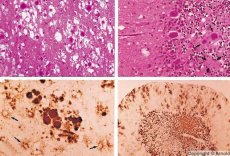
Einige Erreger von Prionenerkrankungen reichern sich zunächst im lymphatischen Gewebe an. Prionen, die ins Gehirn gelangen, reichern sich dort in großen Mengen an und verursachen Amyloidose (extrazelluläre Dysproteinose, gekennzeichnet durch Amyloidablagerung mit Entwicklung von Atrophie und Sklerose des Gewebes) und Astrozytose (Proliferation astrozytischer Neuroglia, Hyperproduktion von Gliafasern). Es bilden sich Fibrillen, Proteinaggregate oder Amyloid sowie spongiforme Veränderungen im Gehirn (transmissible spongiforme Enzephalopathien). Infolgedessen ändern sich das Verhalten, die Bewegungskoordination wird beeinträchtigt und es kommt zu Erschöpfung mit tödlichem Ausgang. Eine Immunität wird nicht ausgebildet. Prionenerkrankungen sind Konformationserkrankungen, die durch eine Fehlfaltung (Verletzung der korrekten Konformation) von zellulärem Protein entstehen, das für die normale Funktion des Körpers notwendig ist. Die Übertragungswege von Prionen sind vielfältig:
- Nahrungsweg – infizierte Produkte tierischen Ursprungs, Lebensmittelzusätze aus rohen Rinderorganen usw.:
- Übertragung durch Bluttransfusionen, Verabreichung von Arzneimitteln tierischen Ursprungs, Organ- und Gewebetransplantation, Verwendung infizierter chirurgischer und zahnärztlicher Instrumente;
- Übertragung durch immunbiologische Präparate (Infektion von 1500 Schafen mit PrP''' durch Brain-Formol-Impfstoff von kranken Schafen ist bekannt).
Pathologische Prionen gelangen nach ihrem Eintritt in den Darm in Blut und Lymphe. Nach peripherer Replikation in Milz, Blinddarm, Mandeln und anderen lymphatischen Geweben werden sie über die peripheren Nerven ins Gehirn transportiert (Neuroinvasion). Prionen können direkt über die Blut-Hirn-Schranke ins Gehirn gelangen. Früher glaubte man, dass sich pathologische Prionen nur im Zentralnervensystem ansammeln, doch Studien haben diese Hypothese widerlegt. Es stellte sich heraus, dass die Ansammlung von Prionen in der Milz mit der Zunahme und Funktion follikulärer dendritischer Zellen einhergeht.
 [
[ Eigenschaften von Prionen
Die normale zelluläre Isoform des Prionproteins mit einem Molekulargewicht von 33–35 kDa wird durch das Prionprotein-Gen bestimmt (das Priongen – PrNP befindet sich auf dem 20. menschlichen Chromosom). Das normale Gen erscheint auf der Zelloberfläche (durch das Glykoprotein des Moleküls in der Membran verankert) und ist empfindlich gegenüber Proteasen. Es reguliert die Übertragung von Nervenimpulsen, Tageszyklen, Oxidationsprozesse, ist am Kupferstoffwechsel im Zentralnervensystem und an der Regulierung der Teilung von Knochenmarksstammzellen beteiligt . Darüber hinaus kommt das Priongen in der Milz, den Lymphknoten, der Haut, dem Gastrointestinaltrakt und den follikulären dendritischen Zellen vor.
Verbreitung pathologischer Prionen
Die Umwandlung von Prionen in veränderte Formen erfolgt, wenn das kinetisch kontrollierte Gleichgewicht zwischen ihnen gestört wird. Dieser Prozess wird durch eine Zunahme der Menge an pathologischen (PrP) oder exogenen Prionen verstärkt. PrP ist ein normales, in der Zellmembran verankertes Protein. PrP' ist ein globuläres, hydrophobes Protein, das mit sich selbst und PrP'' auf der Zelloberfläche Aggregate bildet: Dadurch wandelt sich PrP' in PrP'' um, und der Zyklus setzt sich fort. Die pathologische Form von PrP''' reichert sich in Neuronen an und verleiht der Zelle ein schwammartiges Aussehen.
Kuru
Prionenkrankheit, früher häufig bei den Papua (bedeutet Zittern oder Schütteln) im Osten der Insel Neuguinea. Die infektiösen Eigenschaften der Krankheit wurden von K. Gajdusek nachgewiesen. Der Erreger wird durch Nahrung infolge rituellen Kannibalismus übertragen – durch den Verzehr des unzureichend gekochten, prioneninfizierten Gehirns verstorbener Verwandter. Durch die Schädigung des Zentralnervensystems werden Bewegung und Gang beeinträchtigt, Schüttelfrost und Euphorie („lachender Tod“) treten auf. Die Inkubationszeit beträgt 5–30 Jahre. Der Patient stirbt nach einem Jahr.
Creutzfeldt-Jakob-Krankheit
Prionenerkrankung, die sich als Demenz, Seh- und Kleinhirnstörungen sowie Bewegungsstörungen mit tödlichem Ausgang nach 4-5 Monaten Krankheit bei der klassischen Variante der Creutzfeldt-Jakob-Krankheit und nach 3-14 Monaten bei der neuen Variante der Creutzfeldt-Jakob-Krankheit äußert. Die Inkubationszeit kann 20 Jahre erreichen. Verschiedene Infektionswege und Krankheitsursachen sind möglich:
- beim Verzehr unzureichend wärmebehandelter tierischer Produkte, wie etwa Fleisch und Gehirn von Kühen mit boviner spongiformer Enzephalopathie;
- bei Gewebetransplantationen, wie Hornhauttransplantationen, Bluttransfusionen, der Verwendung von Hormonen und anderen biologisch aktiven Substanzen tierischen Ursprungs, der Verwendung von Katgut, kontaminierten oder unzureichend sterilisierten chirurgischen Instrumenten, prosektoralen Manipulationen;
- im Falle einer Überproduktion von PrR und anderen Bedingungen, die den Prozess der Umwandlung von PrR' in PrR" stimulieren.
Die Krankheit kann auch durch eine Mutation oder Insertion im Prion-Genbereich entstehen. Aufgrund der genetischen Veranlagung zur Creutzfeldt-Jakob-Krankheit ist die Erkrankung häufig familiär. Bei der neuen Variante der Creutzfeldt-Jakob-Krankheit treten die Erkrankungen im jüngeren Alter (durchschnittlich 28 Jahre) auf, im Gegensatz zur klassischen Variante (durchschnittlich 65 Jahre). Bei der neuen Variante der Creutzfeldt-Jakob-Krankheit akkumuliert sich abnormes Prionprotein nicht nur im Zentralnervensystem, sondern auch im lymphoretikulären Gewebe, einschließlich der Mandeln.
Gerstmann-Sträussler-Scheinker-Syndrom
Erbliche Prionenerkrankung, begleitet von Demenz, Hypotonie, Schluckstörungen (Dysphagie) und Dysarthrie. Sie ist häufig familiär bedingt. Die Inkubationszeit beträgt 5 bis 30 Jahre. Die Erkrankung tritt im Alter von 50–60 Jahren auf und dauert 5 bis 13 Jahre.
Erbliche tödliche Schlaflosigkeit
Eine Autoimmunerkrankung mit fortschreitender Schlaflosigkeit, sympathischer Hyperreaktivität (Hypertonie, Hyperthermie, Hyperhidrose, Tachykardie), Tremor, Ataxie, Multiklonen und Halluzinationen. Der Schlaf ist stark gestört. Der Tod tritt mit fortschreitendem Herz-Kreislauf-Versagen ein.
Kratzen
Scrapie (von engl. scrape – kratzen) ist eine Prionenerkrankung der Schafe und Ziegen (Krätze), die mit Schädigungen des zentralen Nervensystems, fortschreitenden Bewegungsstörungen, starkem Hautjucken (Krätze) einhergeht und mit dem Tod des Tieres endet.
Bovine spongiforme Enzephalopathie
Eine Rinderkrankheit, die durch eine Schädigung des Zentralnervensystems, eine gestörte Bewegungskoordination und den unvermeidlichen Tod des Tieres gekennzeichnet ist. Die Epidemie brach erstmals in Großbritannien aus. Sie wurde mit der Fütterung von Tieren mit Fleisch- und Knochenmehl in Verbindung gebracht, das pathologische Prionen enthielt. Die Inkubationszeit beträgt 1,5 bis 15 Jahre. Gehirn, Rückenmark und Augäpfel der Tiere sind am häufigsten infiziert.
Labordiagnostik von Prionenerkrankungen
Bei der Diagnostik werden spongiforme Veränderungen im Gehirn, Astrozytose (Gliose) und das Fehlen entzündlicher Infiltrate festgestellt. Das Gehirn wird auf Amyloid gefärbt. Proteinmarker von Prionenerkrankungen werden im Liquor cerebrospinalis (mittels ELISA) nachgewiesen. Es wird eine genetische Analyse des Prion-Gens (PCR) durchgeführt.
Prävention von Prionenerkrankungen
Zur Dekontamination von Instrumenten und Umgebungsgegenständen werden Autoklavieren (18 min bei 134 °C; 1 h bei 121 °C), Verbrennen, eine zusätzliche Behandlung mit Bleichmittel und einer einnormalen NaCl-Lösung für 1 h empfohlen. Zur unspezifischen Prophylaxe wurden Einschränkungen für die Verwendung von Arzneimitteln tierischen Ursprungs eingeführt, und die Herstellung von Hypophysenhormonen tierischen Ursprungs ist verboten. Die Transplantation der Dura mater ist eingeschränkt. Beim Umgang mit Patientendialyseflüssigkeiten sind Gummihandschuhe zu tragen.