Facharzt des Artikels

Neue Veröffentlichungen
Mukoviszidose
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Mukoviszidose ist eine genetisch bedingte, autosomal-rezessive monogene Erkrankung, die durch eine Störung der Sekretion exokriner Drüsen lebenswichtiger Organe mit Schäden vor allem der Atmungs- und Verdauungssysteme, einem schweren Verlauf und einer ungünstigen Prognose gekennzeichnet ist.
 [ 1 ]
[ 1 ]
Epidemiologie
Die Häufigkeit der Mukoviszidose schwankt zwischen 1:2.500 und 1:4.600 Neugeborenen. Jährlich werden weltweit etwa 45.000 Menschen mit Mukoviszidose geboren. Die Häufigkeit der Mukoviszidose-Genträger beträgt 3–4 %, wobei weltweit etwa 275 Millionen Menschen Träger dieses Gens sind, davon leben etwa 5 Millionen in Russland und etwa 12,5 Millionen in den GUS-Staaten.
Ursachen Mukoviszidose
Mukoviszidose wird autosomal-rezessiv vererbt. Das Mukoviszidose-Gen liegt im Autosom 7, enthält 27 Exons und besteht aus 250.000 Nukleotidpaaren.
Ein einzelnes Gen kann viele Mutationen aufweisen, von denen jede spezifisch für eine bestimmte Population oder geografische Region ist. Mehr als 520 Mutationen wurden beschrieben, die häufigste davon ist Delta-P-508, d. h. ein Austausch der Aminosäure Phenylalanin an Position 508.
Pathogenese
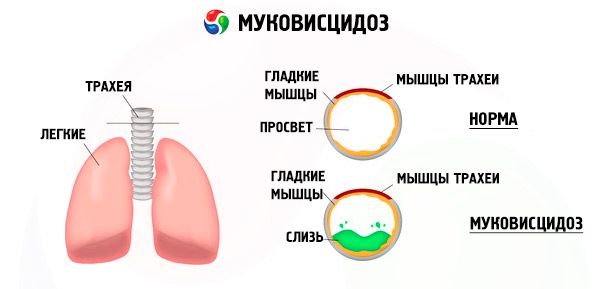
Mutationen im Mukoviszidose-Gen stören die Struktur und Funktion eines Proteins namens CFTR (Cystic Fibrosis Transmembrane Regulator). Dieses Protein fungiert als Chloridkanal und ist am Wasser-Elektrolyt-Austausch von Epithelzellen des bronchopulmonalen Systems, des Magen-Darm-Trakts, der Bauchspeicheldrüse, der Leber und des Fortpflanzungssystems beteiligt. Infolge der Störung der Funktion und Struktur des CFTR-Proteins reichern sich Chloridionen Cl⁻ in der Zelle an. Dies führt zu einer Änderung des elektrischen Potenzials im Lumen der Ausführungsgänge, was den Fluss großer Mengen von Natriumionen (Na⁻) aus dem Lumen des Gangs in die Zelle erleichtert und die Wasseraufnahme aus dem perizellulären Raum weiter verbessert.
Als Folge dieser Veränderungen verdickt sich die Sekretion der meisten exokrinen Drüsen, ihr Abtransport wird gestört, was zu ausgeprägten Folgeerkrankungen der Organe und Systeme führt, am stärksten ausgeprägt im Bronchopulmonal- und Verdauungssystem.
In den Bronchien entwickelt sich ein chronischer Entzündungsprozess unterschiedlicher Intensität, die Funktion des Flimmerepithels ist stark gestört, der Auswurf wird sehr zähflüssig, dickflüssig, sehr schwer abzuführen, seine Stagnation wird beobachtet, es bilden sich Bronchiolo- und Bronchiektasien, die mit der Zeit häufiger werden. Diese Veränderungen führen zu einer Zunahme der Hypoxie und zur Entstehung einer chronischen Lungenherzerkrankung.
Patienten mit Mukoviszidose haben eine hohe Prädisposition für die Entwicklung chronischer Entzündungen im bronchopulmonalen System. Dies ist auf ausgeprägte Störungen des lokalen bronchopulmonalen Abwehrsystems zurückzuführen (verminderte IgA- und Interferonspiegel, verminderte phagozytische Funktion von Alveolarmakrophagen und Leukozyten).
Alveolarmakrophagen spielen eine wichtige Rolle bei der Entstehung chronischer Entzündungen im bronchopulmonalen System. Sie produzieren große Mengen IL-8, was die Chemotaxis neutrophiler Granulozyten im Bronchialbaum drastisch erhöht. Neutrophile akkumulieren in großen Mengen in den Bronchien und sezernieren zusammen mit Epithelzellen zahlreiche proinflammatorische Zytokine, darunter IL-1, 8, 6, Tumornekrosefaktor und Leukotriene.
Eine wichtige Rolle in der Pathogenese von Schäden des bronchopulmonalen Systems spielt auch die hohe Aktivität des Enzyms Elastase. Man unterscheidet zwischen exogener und endogener Elastase. Erstere wird von der Bakterienflora (insbesondere Pseudomonas aeruginosa) produziert, letztere von neutrophilen Leukozyten. Elastase zerstört das Epithel und andere Strukturelemente der Bronchien, was zu weiteren Störungen des mukoziliären Transports und zur schnellen Bildung von Bronchiektasien beiträgt.
Neutrophile Leukozyten sezernieren auch andere proteolytische Enzyme. Alpha-1-Antipyrsin und sekretorische Leukoproteasehemmer wirken dem Einfluss proteolytischer Enzyme entgegen und schützen so das bronchopulmonale System vor deren schädlichen Einflüssen. Leider werden diese Schutzfaktoren bei Patienten mit Mukoviszidose jedoch durch eine signifikante Menge neutrophiler Proteasen unterdrückt.
Alle diese Umstände tragen zur Einführung einer Infektion in das bronchopulmonale System und zur Entwicklung einer chronischen eitrigen Bronchitis bei. Darüber hinaus sollte berücksichtigt werden, dass das vom Mukoviszidose-Gen kodierte defekte Protein den Funktionszustand des Bronchialepithels verändert, was die Adhäsion von Bakterien am Bronchialepithel, vor allem von Pseudomonas aeruginosa, begünstigt.
Neben der Erkrankung des bronchopulmonalen Systems verursacht Mukoviszidose auch schwere Schäden an Bauchspeicheldrüse, Magen, Dick- und Dünndarm sowie Leber.
Symptome Mukoviszidose
Mukoviszidose äußert sich in verschiedenen klinischen Symptomen. Bei Neugeborenen kann sich die Erkrankung durch einen Mekoniumileus äußern. Aufgrund des Mangels oder sogar des völligen Fehlens von Trypsin wird das Mekonium sehr dicht, zähflüssig und akkumuliert im Ileozökalbereich. Es kommt zu einem Darmverschluss, der sich durch starkes Erbrechen mit Gallenbeimischung, Blähungen, fehlende Mekoniumausscheidung, die Entwicklung von Peritonitis-Symptomen und eine rasche Zunahme der klinischen Manifestationen eines schweren Intoxikationssyndroms äußert. Das Kind kann in den ersten Lebenstagen sterben, wenn kein dringender chirurgischer Eingriff durchgeführt wird.
In weniger schweren Fällen ist ein charakteristisches Zeichen der Mukoviszidose reichlicher, häufiger Stuhlgang, fetthaltig, mit viel Fett und sehr unangenehmem Geruch. Bei 1/3 der Patienten wird ein Rektumprolaps beobachtet.
Anschließend leiden die Patienten weiterhin an Darmfunktionsstörungen, einem Malabsorptionssyndrom, schweren körperlichen Entwicklungsstörungen und schwerer Hypovitaminose.
Im ersten oder zweiten Lebensjahr treten Symptome einer Schädigung des bronchopulmonalen Systems (leichte Form der Erkrankung) auf, die sich in einem Husten äußern, der extrem ausgeprägt sein kann und einem Keuchhusten ähnelt. Der Husten wird von Zyanose, Kurzatmigkeit und der Absonderung von dickem, zunächst schleimigem, dann eitrigem Auswurf begleitet. Allmählich bildet sich ein klinisches Bild von chronisch obstruktiver Bronchitis und Bronchiektasie, Lungenemphysem und Atemversagen. Kinder sind extrem anfällig für akute virale und bakterielle Infektionen der Atemwege, was zu Exazerbationen und zum Fortschreiten der bronchopulmonalen Pathologie beiträgt. Die Entwicklung eines infektionsabhängigen Asthma bronchiale ist möglich.
Bei Kindern im Schulalter kann sich Mukoviszidose als „Darmkolik“ äußern. Patienten klagen über starke paroxysmale Bauchschmerzen, Blähungen und wiederholtes Erbrechen. Beim Abtasten des Bauches werden dichte Formationen festgestellt, die sich in der Projektion des Dickdarms befinden – Stuhlmassen, vermischt mit dickem, dichtem Schleim. Kinder sind sehr anfällig für die Entwicklung einer hypochlorämischen Alkalose aufgrund übermäßiger Salzausscheidung mit Schweiß bei heißem Wetter, während sich auf der Haut des Kindes „Salzfrost“ bildet.
Erkrankungen des bronchopulmonalen Systems bei Erwachsenen
Schäden am bronchopulmonalen System bei Patienten mit Mukoviszidose (pulmonale Form der Erkrankung) sind durch die Entwicklung einer chronischen eitrigen obstruktiven Bronchitis, Bronchiektasien, chronischer Lungenentzündung, Lungenemphysem, Atemversagen und Lungenherzerkrankung gekennzeichnet. Einige Patienten entwickeln einen Pneumothorax und andere Komplikationen der Mukoviszidose: Atelektase, Lungenabszesse, Hämoptyse, Lungenblutung und infektionsabhängiges Asthma bronchiale.
Die Patienten klagen über einen schmerzhaften paroxysmalen Husten mit sehr zähflüssigem, schwer abtrennbarem schleimig-eitrigem Auswurf, manchmal mit einer Beimischung von Blut. Darüber hinaus ist Kurzatmigkeit äußerst charakteristisch, zunächst bei körperlicher Anstrengung und dann in Ruhe. Kurzatmigkeit wird durch eine Bronchialobstruktion verursacht. Viele Patienten klagen über chronische Rhinitis, verursacht durch Polyposis und Sinusitis. Charakteristisch sind ebenfalls erhebliche Schwäche, fortschreitender Leistungsabfall und häufige akute Viruserkrankungen der Atemwege. Bei der Untersuchung fallen blasse Haut, Schwellungen im Gesicht, Zyanose der sichtbaren Schleimhäute und starke Kurzatmigkeit auf. Mit der Entwicklung einer dekompensierten pulmonalen Herzkrankheit treten Ödeme in den Beinen auf. Eine Verdickung der Endphalangen der Finger in Form von Trommelschlägeln und Nägeln in Form von Uhrgläsern kann beobachtet werden. Der Brustkorb nimmt eine tonnenförmige Form an (aufgrund der Entwicklung eines Lungenemphysems).
Die Perkussion der Lunge zeigt Anzeichen eines Emphysems – ein kastenförmiges Geräusch, eine starke Einschränkung der Beweglichkeit des Lungenrandes und eine Absenkung des unteren Lungenrandes. Die Auskultation der Lunge zeigt eine schwere Atmung mit verlängerter Ausatmung, vereinzeltes trockenes Keuchen sowie feuchtes, mittleres und feines, sprudelndes Keuchen. Bei einem schweren Lungenemphysem ist die Atmung stark geschwächt.
Extrapulmonale Manifestationen der Mukoviszidose
Extrapulmonale Manifestationen der Mukoviszidose können sehr ausgeprägt sein und treten häufig auf.
Pankreasschäden
Bei 85 % der Patienten mit Mukoviszidose wird eine Insuffizienz der exokrinen Funktion der Bauchspeicheldrüse unterschiedlichen Schweregrades beobachtet. Bei geringfügigen Pankreasschäden fehlen Maldigestion und Malabsorptionssyndrome, es gibt lediglich Labormanifestationen einer exokrinen Insuffizienz (niedrige Trypsin- und Lipasewerte im Blut und Duodenalinhalt; oft schwere Steatorrhoe). Es ist bekannt, dass zur Vorbeugung eines Maldigestion-Syndroms die Sekretion von nur 1 bis 2 % der Gesamtlipase ausreicht. Klinisch manifestieren sich nur signifikante Störungen der exokrinen Funktion.
Unter normalen Bedingungen produzieren die Azini der Bauchspeicheldrüse ein flüssiges, enzymreiches Sekret. Auf ihrem Weg durch den Ausführungsgang reichert sich das Sekret mit Wasser und Anionen an und wird noch flüssiger. Bei Mukoviszidose erhält das Pankreassekret aufgrund einer Störung der Struktur und Funktion des Transmembranregulators (Chloridkanal) nicht genügend Flüssigkeit, wird zähflüssig und seine Bewegung durch den Ausführungsgang verlangsamt sich stark. Die Proteine des Sekrets lagern sich an den Wänden der kleinen Ausführungsgänge ab und verstopfen diese. Mit fortschreitender Erkrankung kommt es schließlich zur Zerstörung und Atrophie der Azini – es entsteht eine chronische Pankreatitis mit exokriner Pankreasinsuffizienz. Klinisch spiegelt sich dies in der Entwicklung von Maldigestion- und Malabsorptionssyndromen wider. Pankreasinsuffizienz ist die Hauptursache für Fettmalabsorption bei Mukoviszidose, die jedoch meist mit einem signifikanten Lipasemangel einhergeht. Forsher und Durie (1991) weisen darauf hin, dass bei völliger Abwesenheit von Pankreaslipase Fett zu 50–60 % abgebaut und resorbiert wird. Dies ist auf das Vorhandensein von Magen- und Speichellipasen (sublingual) zurückzuführen, deren Aktivität nahe der unteren Normgrenze liegt. Neben der Störung des Fettabbaus und der Fettresorption kommt es auch zu einer Störung des Proteinabbaus und der Proteinresorption. Etwa 50 % des mit der Nahrung aufgenommenen Proteins gehen mit dem Kot verloren. Die Kohlenhydrataufnahme ist trotz des α-Amylasemangels weniger beeinträchtigt, der Kohlenhydratstoffwechsel kann jedoch erheblich gestört sein.
Eine Schädigung der Bauchspeicheldrüse äußert sich in der Entwicklung eines Maldigestion- und Malabsorptionssyndroms mit erheblichem Gewichtsverlust und reichlich fettem Stuhl.
Die Entwicklung von Maldigestion- und Malabsorptionssyndromen wird auch durch eine schwere Funktionsstörung der Darmdrüsen, eine gestörte Darmsaftsekretion und eine Abnahme des Gehalts an Darmenzymen begünstigt.
Maldigestion- und Malabsorptionssyndrome werden auch als intestinale Form der Mukoviszidose bezeichnet.
Eine beeinträchtigte endokrine Funktion der Bauchspeicheldrüse (Diabetes mellitus) wird bei Patienten mit Mukoviszidose im Spätstadium der Erkrankung beobachtet (bei 2 % der Kinder und 15 % der Erwachsenen).
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Leber- und Gallenwegsschäden
Bei 13 % der Patienten mit gemischten und intestinalen Formen der Mukoviszidose entwickelt sich eine Leberzirrhose. Sie ist am typischsten für die Mutationen W128X, Delta-P508 und X1303K. Eine biliäre Leberzirrhose mit portaler Hypertonie wird bei 5-10 % der Patienten festgestellt. Laut Welch, Smith (1995) werden bei 86 % der Patienten mit Mukoviszidose klinische, morphologische, laborchemische und instrumentelle Anzeichen einer Leberschädigung festgestellt.
Viele Patienten mit Mukoviszidose entwickeln auch eine chronische Cholezystitis, oft mit Steinen.
Funktionsstörung der Geschlechtsdrüsen
Bei Patienten mit Mukoviszidose kann es zu Azoospermie kommen, die die Ursache für Unfruchtbarkeit ist. Auch bei Frauen ist eine verminderte Fruchtbarkeit typisch.
Bühnen
Es gibt drei Schweregrade der pulmonalen Mukoviszidose.
Die leichte Form der Mukoviszidose ist durch seltene Exazerbationen gekennzeichnet (nicht mehr als einmal pro Jahr); während der Remissionsphasen treten praktisch keine klinischen Manifestationen auf und die Patienten sind arbeitsfähig.
Mäßiger Schweregrad - Exazerbationen werden 2-3 Mal pro Jahr beobachtet und dauern etwa 2 Monate oder länger. In der Exazerbationsphase kommt es zu starkem Husten mit schwer absonderbarem Auswurf, Kurzatmigkeit auch bei geringer körperlicher Anstrengung, subfebriler Körpertemperatur, allgemeiner Schwäche und Schwitzen. Gleichzeitig liegt eine Verletzung der exokrinen Funktion der Bauchspeicheldrüse vor. In der Remissionsphase ist die Arbeitsfähigkeit nicht vollständig wiederhergestellt, Kurzatmigkeit bei körperlicher Anstrengung bleibt bestehen.
Ein schwerer Verlauf ist durch sehr häufige Exazerbationen der Erkrankung gekennzeichnet. Remissionen treten praktisch nicht auf. Im klinischen Bild treten schweres Atemversagen und Symptome einer chronischen pulmonalen Herzerkrankung, häufig dekompensiert, typisch für Hämoptyse, in den Vordergrund. Es kommt zu einem signifikanten Gewichtsverlust, die Patienten sind vollständig behindert. Eine schwere bronchopulmonale Pathologie geht in der Regel mit einer ausgeprägten Pankreasfunktionsstörung einher.
Formen
- Bronchopulmonale Läsionen
- Wiederholte und wiederkehrende Lungenentzündung mit langwierigem Verlauf.
- Abszesspneumonie, insbesondere bei Säuglingen.
- Chronische Lungenentzündung, insbesondere beidseitig.
- Asthma bronchiale, das auf herkömmliche Therapie nicht anspricht.
- Rezidivierende Bronchitis, Bronchiolitis, insbesondere bei Pseudomonas aeruginosa-Kultur.
- Veränderungen im Magen-Darm-Trakt
- Mekoniumileus und seine Äquivalente.
- Syndrom der gestörten intestinalen Resorption unbekannter Genese.
- Verschlussikterus beim Neugeborenen mit langwierigem Verlauf.
- Leberzirrhose.
- Diabetes mellitus.
- Gastroösophagealer Reflux.
- Cholelithiasis.
- Rektumprolaps.
- Veränderungen in anderen Organen und Systemen
- Wachstums- und Entwicklungsstörungen.
- Verzögerte sexuelle Entwicklung.
- Männliche Unfruchtbarkeit.
- Nasenpolypen.
- Geschwister aus Familien mit Mukoviszidose.
[ 24 ]
Komplikationen und Konsequenzen
Zu den Komplikationen des Magen-Darm-Trakts zählen:
- Bei 8–12 % der Patienten über 25 Jahren entwickelt sich Diabetes mellitus.
- Fibrosierende Kolonopathie.
- Mekoniumileus in der Neugeborenenperiode (bei 12 % der Neugeborenen mit Mukoviszidose), distales Darmverschlusssyndrom, Rektumprolaps, Magengeschwüre und gastroösophageale Refluxkrankheit.
Leberkomplikationen:
- Fettlebererkrankung (bei 30-60 % der Patienten),
- Fokale biliäre Zirrhose, multinoduläre biliäre Zirrhose und damit verbundene portale Hypertonie.
Portale Hypertonie führt aufgrund von Ösophagusvarizen manchmal zum Tod.
Bei Patienten mit Mukoviszidose kommt es häufiger zu Cholezystitis und Gallensteinen als bei anderen Personen.
Verzögerte Pubertät und verminderte Fruchtbarkeit sowie andere Komplikationen. Die meisten Männer leiden an Azoospermie und einer Unterentwicklung der Samenleiter.
Diagnose Mukoviszidose
Allgemeine Blutanalyse – Typisch ist eine Anämie unterschiedlichen Schweregrades, meist normo- oder hypochrom. Die Anämie hat eine polyfaktorielle Genese (verminderte Aufnahme von Eisen und Vitamin B12 im Darm aufgrund der Entwicklung eines Malabsorptionssyndroms). Leukopenie ist möglich, mit der Entwicklung einer eitrigen Bronchitis und Lungenentzündung – Leukozytose, erhöhte BSG.
Allgemeine Urinanalyse – keine signifikanten Veränderungen, in seltenen Fällen wird eine leichte Proteinurie beobachtet.
Koprologische Untersuchung – Steatorrhoe, Creatorrhoe werden beobachtet. Becker (1987) empfiehlt die Messung von Chymotrypsin und Fettsäuren im Stuhl. Vor der Bestimmung von Chymotrypsin im Stuhl ist es notwendig, die Einnahme von Verdauungsenzymen mindestens 3 Tage vor der Untersuchung abzusetzen. Bei Mukoviszidose ist die Chymotrypsinmenge im Stuhl reduziert und die Menge an Fettsäuren erhöht (die normale Ausscheidung von Fettsäuren beträgt weniger als 20 mmol/Tag). Es ist zu berücksichtigen, dass eine erhöhte Ausscheidung von Fettsäuren mit dem Stuhl auch beobachtet wird bei:
- Mangel an konjugierten Fettsäuren im Dünndarm aufgrund von Leberversagen, Verstopfung der Gallenwege, erheblicher bakterieller Besiedlung des Dünndarms (in diesem Fall kommt es zu einer intensiven Hydrolyse der Gallensäuren);
- Ileitis;
- Zöliakie (mit der Entwicklung eines Malabsorptionssyndroms);
- Enteritis;
- Darmlymphome;
- Whipple-Krankheit;
- Nahrungsmittelallergien;
- beschleunigter Transit von Nahrungsmassen bei Durchfall unterschiedlicher Herkunft, Karzinoid-Syndrom, Thyreotoxikose.
Biochemischer Bluttest – verringerte Gesamtprotein- und Albuminwerte, erhöhte Werte von Alpha2- und Gammaglobulinen, Bilirubin und Aminotransferasen (im Falle einer Leberschädigung), verringerte Aktivität von Amylase, Lipase, Trypsin sowie Eisen- und Kalziumwerte (im Falle der Entwicklung eines Maldigestion-Syndroms, Malabsorption).
Sputumanalyse – Vorhandensein einer großen Anzahl neutrophiler Leukozyten und Mikroorganismen (bei der Sputumbakterioskopie).
Eine Untersuchung der Resorptionsfunktion des Dünndarms und der exokrinen Funktion der Bauchspeicheldrüse zeigt erhebliche Störungen.
Röntgenuntersuchung der Lunge - zeigt Veränderungen, deren Schweregrad von der Schwere und Phase der Erkrankung abhängt. Die charakteristischsten Veränderungen sind:
- erhöhte Lungenmusterung aufgrund peribronchialer interstitieller Veränderungen;
- Erweiterung der Lungenwurzeln;
- Bild einer lobulären, subsegmentalen oder auch segmentalen Atelektase der Lunge;
- erhöhte Transparenz der Lungenfelder, vor allem in den oberen Abschnitten, niedrige Position und unzureichende Beweglichkeit des Zwerchfells, Erweiterung des retrosternalen Raums (Manifestation eines Lungenemphysems);
- segmentale oder polysegmentale Infiltration des Lungengewebes (bei der Entwicklung einer Lungenentzündung).
Die Bronchographie zeigt Veränderungen, die durch eine Verstopfung der Bronchien durch zähflüssigen Auswurf verursacht werden (Fragmentierung der Bronchienfüllung mit Kontrastmittel, ungleichmäßige Konturen, das Phänomen der Bronchialruptur, eine signifikante Abnahme der Anzahl der Seitenäste) sowie Bronchoekgase (zylindrisch, gemischt), die hauptsächlich in den unteren Teilen der Lunge lokalisiert sind.
Bei der Bronchoskopie zeigt sich eine diffuse eitrige Bronchitis mit reichlich dickem, zähflüssigem Auswurf und fibrinösen Belägen.
Spirometrie – bereits in den frühen Stadien der Erkrankung zeigt sich eine Ateminsuffizienz vom obstruktiven Typ (verminderter FVC, FEV1, Tiffno-Index), restriktiven (verminderter FVC) oder am häufigsten obstruktiv-restriktive (verminderter FVC, FVC, FEV1, Tiffno-Index).
Der Gibson-Cook-Schweißtest (Schweißelektrolyttest) ist eine Stimulation des Schwitzens mittels Pilocarpin-Elektrophorese mit anschließender Bestimmung der Chloride im Schweiß. Doerehuk (1987) beschreibt den Test wie folgt. Die Pilocarpin-Elektrophorese wird am Unterarm durchgeführt, die Stromstärke beträgt 3 mA. Nach der Reinigung der Haut mit destilliertem Wasser wird der Schweiß mithilfe von Filterpapier gesammelt, das auf die stimulierte Stelle gelegt und mit Gaze abgedeckt wird, um Verdunstung zu verhindern. Nach 30–60 Minuten wird das Filterpapier entfernt und in destilliertem Wasser eluiert. Die gesammelte Schweißmenge wird gemessen. Für zuverlässige Ergebnisse müssen mindestens 50 mg (vorzugsweise 100 mg) Schweiß gesammelt werden.
Bei einem Chloridgehalt über 60 mmol/l gilt die Diagnose Mukoviszidose als wahrscheinlich, bei einem Chloridgehalt über 100 mmol/l als sicher; in diesem Fall sollte die Differenz der Chlor- und Natriumkonzentrationen 8–10 mmol/l nicht überschreiten. Hadson (1983) empfiehlt bei grenzwertigen Natrium- und Chloridwerten im Schweiß einen Prednisolontest (5 mg oral über 2 Tage, anschließend Bestimmung der Elektrolyte im Schweiß). Bei Personen ohne Mukoviszidose sinkt der Natriumspiegel im Schweiß bis an die untere Normgrenze; bei Mukoviszidose ändert er sich nicht. Ein Schweißtest wird bei jedem Kind mit chronischem Husten empfohlen.
Die Analyse von Blutproben oder DNA-Proben auf wichtige Mutationen des Mukoviszidose-Gens ist der empfindlichste und spezifischste diagnostische Test. Diese Methode eignet sich jedoch nur für Länder, in denen die Delta-P508-Mutationsrate über 80 % liegt. Zudem ist die Technik sehr teuer und technisch aufwendig.
Die pränatale Diagnose von Mukoviszidose erfolgt durch Bestimmung der Isoenzyme der alkalischen Phosphatase im Fruchtwasser. Diese Methode ist ab der 18. bis 20. Schwangerschaftswoche möglich.
Die wichtigsten Kriterien für die Diagnose von Mukoviszidose sind die folgenden:
- Hinweise in der Anamnese auf eine verzögerte körperliche Entwicklung im Kindesalter, wiederkehrende chronische Atemwegserkrankungen, dyspeptische Störungen und Durchfall, das Vorliegen einer Mukoviszidose bei nahen Verwandten;
- chronisch obstruktive Bronchitis, oft rezidivierend, mit der Entwicklung von Bronchiektasien und Lungenemphysem, oft rezidivierende Lungenentzündung;
- chronisch rezidivierende Pankreatitis mit deutlicher Abnahme der exokrinen Funktion, Malabsorptionssyndrom;
- erhöhter Chlorgehalt im Schweiß des Patienten;
- Unfruchtbarkeit mit erhaltener Sexualfunktion.
Eine erfolgreiche Diagnose und Differenzialdiagnose der Mukoviszidose wird durch die Identifizierung von Risikogruppen erleichtert.
Mukoviszidose-Screening-Programm
- Allgemeine Analyse von Blut, Urin, Auswurf.
- Bakteriologische Analyse des Auswurfs.
- Koprologische Analyse.
- Biochemische Blutuntersuchung: Bestimmung von Gesamtprotein und Proteinfraktionen, Glucose, Bilirubin, Aminotransferasen, alkalischer Phosphatase, Gamma-Glutamyl-Transpeptidasen, Kalium, Calcium, Eisen, Lipase, Amylase, Trypsin.
- Untersuchung der exokrinen Funktion der Bauchspeicheldrüse und der Absorptionsfunktion des Darms.
- Durchleuchtung und Röntgen der Lunge, CT-Scan der Lunge.
- EKG.
- Echokardiographie.
- Bronchoskopie und Bronchographie.
- Spirometrie.
- Schweißtest.
- Beratung durch einen Genetiker.
- Analyse von Blutproben oder DNA-Proben auf schwerwiegende Mutationen des Mukoviszidose-Gens.
Was muss untersucht werden?
Welche Tests werden benötigt?
Wen kann ich kontaktieren?
Behandlung Mukoviszidose
Art und Schwere der Symptome der Mukoviszidose können sehr unterschiedlich sein, daher gibt es keinen typischen Behandlungsplan; die Behandlung ist auf jeden Patienten individuell zugeschnitten.
Die Therapie besteht aus folgenden therapeutischen Maßnahmen:
- Atemübungen und Lagerungsdrainage helfen, zähen Schleim, der sich in der Lunge ansammelt, zu entfernen. Einige Techniken zur Atemwegsbefreiung erfordern die Unterstützung von Familienmitgliedern, Freunden oder einem Lungenfacharzt. Viele Patienten verwenden eine aufblasbare Brustweste, die mit hoher Frequenz vibriert.

- Inhalationsmedikamente mit bronchodilatatorischer, entwässernder (Mukolytika) und antibakterieller Wirkung (zum Beispiel Fluorchinolone).
- Präparate mit Pankreasenzymen zur Verbesserung der Verdauung. Diese Präparate werden zu den Mahlzeiten eingenommen.
- Multivitamine (einschließlich fettlöslicher Vitamine).
Im Jahr 2015 genehmigte die FDA ein zweites Medikament zur Behandlung von Mukoviszidose, das auf ein defektes Protein namens CFTR abzielt. Das erste Medikament, ein sogenannter CFTR-Modulator, wurde 2012 zugelassen. CFTR-Modulatoren sollen das Leben mancher Mukoviszidose-Patienten um Jahrzehnte verlängern.
Zur Behandlung der folgenden Atemwegskomplikationen kann eine Operation erforderlich sein:
- Pneumothorax, massive wiederkehrende oder anhaltende Hämoptyse, Nasenpolypen, anhaltende und chronische Sinusitis.
- Mekoniumileus, Intussuszeption, Rektumprolaps.
Im Endstadium der Erkrankung wird eine Lungentransplantation durchgeführt.
Prognose
Das durchschnittliche Überlebensalter von Patienten mit Mukoviszidose liegt zwischen 35 und 40 Jahren. Männer sind dabei höher als Frauen.
Mit modernen Behandlungsstrategien erreichen 80 % der Patienten das Erwachsenenalter. Allerdings schränkt Mukoviszidose die funktionellen Fähigkeiten der Patienten erheblich ein. Eine Heilung für diese Krankheit ist noch immer nicht möglich.