Facharzt des Artikels

Neue Veröffentlichungen
Tollwut bei Kindern
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Tollwut oder Tollwut ist eine akute Viruserkrankung, die durch den Biss eines infizierten Tieres übertragen wird und zu Schäden am Nervensystem und der Entwicklung einer schweren Enzephalitis mit tödlichem Ausgang führt.
Epidemiologie
Das Tollwutvirus ist seit der Antike eine Plage für die öffentliche Gesundheit und verursacht derzeit jährlich etwa 59.000 Todesfälle, die fast ausschließlich durch Hundebisse übertragen werden. Dies hat erhebliche wirtschaftliche Auswirkungen auf Entwicklungsländer, insbesondere in Afrika und Asien, die solche Verluste am wenigsten verkraften können. Trotz der fast 100%igen Sterblichkeitsrate ist die Hundetollwut jedoch eine vollständig vermeidbare Krankheit, wie historische Beispiele für die Ausrottung der Hundetollwut in Industrieländern belegen. [ 1 ]
Ursachen Tollwut
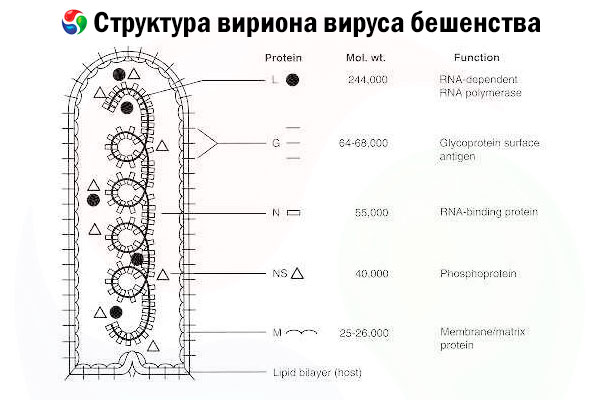
Der Erreger ist das Tollwutvirus (RV), ein Negativstrang-RNA-Virus aus der Familie der Rhabdoviren mit einer Größe von etwa 60 nm × 180 nm.
Es besteht aus einem inneren Proteinkern (Nukleokapsid), der Nukleinsäure enthält, und einer äußeren Membran, einer lipidhaltigen Doppelschicht, die mit transmembranären Glykoprotein-Spikes bedeckt ist. Es hat eine relativ einfache modulare Genomstruktur und kodiert fünf Strukturproteine:
- RNA-abhängige RNA-Polymerase (L),
- Nukleoprotein (N),
- phosphoryliertes Protein (P),
- Matrixprotein (M) und
- äußeres Oberflächenglykoprotein (G).
Die Proteine N, P und L bilden zusammen mit der genomischen RNA den Ribonukleoproteinkomplex. G ist das einzige RV-Antigen, das die Produktion von RV-neutralisierenden Antikörpern induzieren kann, den wichtigsten Immuneffektoren gegen eine letale RV-Infektion. Andererseits hat sich der Ribonukleoproteinkomplex als das wichtigste RV-Antigen erwiesen, das CD4+-T-Zellen induzieren kann, welche die Produktion von RV-neutralisierenden Antikörpern durch intrastrukturelle Antigenerkennung steigern können.[ 2 ] Der Ribonukleoproteinkomplex könnte eine wichtige Rolle bei der Etablierung des immunologischen Gedächtnisses und der Langzeitimmunität spielen.[ 3 ]
 [
[ Klassifizierung und Antigentypen
Die Gattung Lyssavirus umfasst das Tollwutvirus und antigenisch und genetisch verwandte Tollwutviren: Lagos-, Mokola- und Duvenhage-Fledermausviren sowie zwei mutmaßliche Subtypen europäischer Fledermauslyssaviren. Kreuzimmunitätsstudien deuten darauf hin, dass Tiere, die mit herkömmlichen Tollwutimpfstoffen immunisiert wurden, bei einer Infektion mit anderen Lyssaviren möglicherweise nicht vollständig geschützt sind.
Tollwutviren lassen sich als fixierte (durch Passage in Tieren oder Zellkulturen adaptierte) oder Straßenviren (Wildtyp) klassifizieren. Der Einsatz monoklonaler Antikörper und genetischer Sequenzierung zur Unterscheidung von Straßen-Tollwutviren hat dazu beigetragen, Virusvarianten aus wichtigen Wirtsreservoirs weltweit zu identifizieren und mögliche menschliche Expositionsquellen zu identifizieren, wenn bei einem Patienten ansonsten kein eindeutiger Tierbiss nachgewiesen werden konnte.[ 8 ]
Pathogenese
Das Hauptreservoir und die Hauptinfektionsquelle bei Wildtieren sind Wölfe, Füchse, Schakale und Fledermäuse. Bei Haustieren – Hunden und Katzen, selten – sind Pferde, Rinder, Schweine, Ratten usw. Die Übertragung der Infektion von Mensch zu Mensch ist zwar möglich, aber äußerst selten. Dies ist eine typische Zoonose. Menschen werden hauptsächlich von Hunden mit Tollwut infiziert.
Nach einem Biss eines Menschen durch ein krankes Tier vermehrt sich das Virus im Muskelgewebe an der Bissstelle. Nachdem es die Enden der sensorischen peripheren Nerven erreicht hat, breitet es sich zentripetal aus und erreicht die Motoneuronen. Die Zeit, die das Virus benötigt, um sich auszubreiten und das Gehirn zu befallen, hängt von der Bissstelle ab. Bei schweren Bissen an Kopf und Gesicht kann das Virus das zentrale Nervensystem innerhalb von 15 bis 20 Tagen erreichen. Bei leichten Verletzungen der Haut an Rumpf und Gliedmaßen und einer daraus resultierenden geringen Erregerdosis kann sich die Ausbreitung des Virus in das zentrale Nervensystem um mehrere Monate oder sogar bis zu ein bis anderthalb Jahre verzögern. Im zentralen Nervensystem angekommen, fixiert sich das Virus im Gewebe von Gehirn und Rückenmark, hauptsächlich in den Neuronen der Medulla oblongata, des Ammonshorns und der Hirnbasis. Im Rückenmark sind die Hinterhörner am stärksten betroffen. Vom zentralen Nervensystem gelangt das Virus zentrifugal entlang der Nervenstämme in die Speicheldrüsen, wo es sich vermehrt und mit dem Speichel ausgeschieden wird.
Konzepte zur Pathogenese der Tollwut
RV hat ein breites Wirtsspektrum und kann fast alle Säugetiere infizieren. Obwohl verschiedene Übertragungswege für RV beschrieben wurden, erfolgt die natürliche Infektion meist durch einen Biss. Neben Bissen kann der Verzehr von RV-infizierten Kadavern eine Tollwutvirusinfektion bei Polarfüchsen fördern, und der Kontakt von RV mit Schleimhäuten ist ein weiterer möglicher Übertragungsweg.[ 9 ] Unter ungewöhnlichen Umständen, wie z. B. der versehentlichen Freisetzung von RV als Aerosol in einem Labor oder als Aerosol in Höhlen, die von vielen Fledermäusen bewohnt werden,[ 10 ] kann es zu einer Aerosolübertragung kommen.
Es ist noch nicht klar, ob sich Straßen-RV und an Mäuse oder Gewebekulturen adaptierte RV-Stämme an der Inokulationsstelle replizieren, bevor sie ins ZNS gelangen. Während experimentelle intramuskuläre Infektionen junger Hamster oder Waschbären mit Straßen-RV eine RV-Replikation in quergestreiften Muskelzellen zeigten, bevor das Virus über neuromuskuläre Verbindungen in die Axone der Motoneuronen eindrang,[ 11 ][ 12 ] zeigte die intramuskuläre Infektion von Mäusen mit an Mäuse adaptiertem CVS-24 RV, dass RV ohne vorherige Replikation an der Inokulationsstelle direkt ins ZNS wandert.[ 13 ] In den Enden der unmyelinierten Axone angekommen, wird RV retrograd zum Zellkörper transportiert.
Neuere Erkenntnisse deuten darauf hin, dass der axonale Vesikeltransport eine Schlüsselstrategie für die Fernbewegung von Virionen in Axonen darstellt.[ 14 ] Schätzungsweise wandert RV innerhalb von Axonen mit einer Geschwindigkeit von 3 mm/h.[ 15 ] Die Infektion breitet sich dann über eine Kette von Neuronen aus, die durch synaptische Verbindungen miteinander verbunden sind. Der genaue Mechanismus, der die transsynaptische Ausbreitung fördert, ist jedoch noch unbekannt. Nach der Infektion des Gehirns breitet sich das Virus zentrifugal im peripheren und autonomen Nervensystem vieler peripherer Organe aus.[ 16 ] Im letzten Stadium des Infektionszyklus wandert RV zu den Speicheldrüsen; nach der Replikation in mukogenen Azinuszellen wird es in den Speichel freigesetzt und ist bereit für die Übertragung auf den nächsten Wirt.[ 17 ]
Im Hinblick auf die durch das Tollwutvirus induzierte Pathologie wurde apoptotischer Zelltod als möglicher pathogener Mechanismus in experimentellen Tollwutmodellen von Mäusen vorgeschlagen, die mit einem fixierten Stamm von RV infiziert waren.[ 18 ] Ein pathogener Mechanismus, der zu der für Tollwut charakteristischen schweren Funktionsstörung des ZNS beitragen könnte, ist möglicherweise eine beeinträchtigte neuronale Funktion. Es zeigte sich, dass die Genexpression in RV-infizierten Neuronen deutlich verringert ist, was zu einer allgemeinen Unterdrückung der Proteinsynthese führt,[ 19 ] und mehrere Studien zeigten eine beeinträchtigte Neurotransmission nach einer RV-Infektion. Jiang zeigte, dass die Bindung eines Acetylcholinrezeptor-Antagonisten an infizierte Rattenhirnhomogenate im Vergleich zu Kontrollen verringert war.[ 20 ] Auch eine beeinträchtigte Freisetzung und Bindung von Serotonin, einem Neurotransmitter, der an der Kontrolle von Schlafzyklus, Schmerzwahrnehmung und Verhalten beteiligt ist, wurde im RV-infizierten Rattenhirn beobachtet. [ 21 ], [ 22 ] Neben der Beeinflussung der Neurotransmission kann eine Infektion des rechten Ventrikels auch die Ionenkanäle beeinträchtigen. Infizierte Neuroblastomzellen der Maus weisen eine verminderte funktionelle Expression spannungsgesteuerter Natriumkanäle auf, was Aktionspotentiale verhindern und letztlich zu einer Funktionsbeeinträchtigung führen kann. [ 23 ]
Zusätzlich zum Fehlen schwerer pathologischer Läsionen im ZNS lösen die meisten Fälle von Tollwut beim Menschen 7 bis 10 Tage nach dem Einsetzen der klinischen Symptome keine Immunreaktion aus. Diese schwerwiegenden Unterschiede zwischen der Pathogenese der Tollwut und der der meisten anderen viralen oder bakteriellen ZNS-Infektionen werden zusätzlich durch die Tatsache unterstützt, dass eine Immunsuppression entweder wirkungslos ist oder sich nachteilig auf den Ausgang der Tollwut auswirkt. [ 24 ] Die bei Tollwutopfern oft beobachtete niedrige Immunreaktion ist rätselhaft, da sie nicht durch die geringe Immunogenität der RV-Antigene erklärt werden kann. Tatsächlich sind RV-G und das Nukleokapsidprotein potente B- und T-Zell-Antigene, wenn sie parenteral verabreicht werden. [ 25 ] Eine mögliche Erklärung für die niedrige Immunreaktion gegen RV bei Menschen oder Tieren mit Tollwut könnte sein, dass die RV-Infektion des ZNS eine Immunsuppression verursacht, [ 26 ] und es wurde vorgeschlagen, dass RV eine subversive Strategie verfolgt, die die Verhinderung der Apoptose und die Zerstörung eindringender T-Zellen einschließt. [ 27 ]
Abgeschwächte RV-Stämme, die an nicht-neuronale Zellen adaptiert wurden, unterscheiden sich signifikant von pathogenen Straßen-RV-Stämmen in ihrer Neuroinvasivität, d. h. ihrer Fähigkeit, von peripheren Stellen in das ZNS einzudringen. In dieser Hinsicht haben an Gewebekulturen adaptierte RV-Stämme entweder keine oder nur eine eingeschränkte Fähigkeit, von peripheren Stellen in das ZNS einzudringen, während Straßen-RV-Stämme oder an Mäuse adaptierte RV-Stämme wie CVS-24 hochinvasiv sind.[ 28 ] Zu den Schlüsselfaktoren der RV-Neuroinvasion gehören die Virusaufnahme, der axonale Transport, die transsynaptische Ausbreitung und die Virusreplikationsrate.
Bis vor Kurzem war unser Wissen über die Pathogenese von RV begrenzt und basierte hauptsächlich auf deskriptiven Studien an Straßen-RV-Stämmen oder experimentellen Infektionen mit im Labor adaptierten, abgeschwächten Stämmen. Dank der Reverse-Genetik-Technologie konnten wir die viralen Elemente identifizieren, die den pathogenen Phänotyp von RV bestimmen, und die Mechanismen der Tollwut-Pathogenese besser verstehen.
Identifizierung viraler Elemente, die die Aufnahme, Verbreitung und Replikation des Tollwutvirus kontrollieren
- Virale Elemente, die an der Virenerfassung beteiligt sind
Die RV-Infektion beginnt mit der Anheftung des Virus an einen mutmaßlichen zellulären Rezeptor. Obwohl mehrere Membranoberflächenmoleküle als RV-Rezeptoren vorgeschlagen wurden, darunter der nikotinische Acetylcholinrezeptor[ 29 ], das neuronale Zelladhäsionsmolekül[ 30 ] und der niedrigaffine Neurotrophinrezeptor p75 NTR[ 31 ], ist noch unklar, ob diese Moleküle tatsächlich eine Rolle im Lebenszyklus des Tollwutvirus spielen. In diesem Zusammenhang wurde kürzlich gezeigt, dass die RV G–p75 NTR-Interaktion für die RV-Infektion primärer Neuronen nicht erforderlich ist.[ 32 ] Nach der Rezeptorbindung wird RV durch adsorptive oder rezeptorvermittelte Endozytose internalisiert.[ 33 ] Der niedrige pH-Wert im endosomalen Kompartiment induziert dann Konformationsänderungen in RV G, die die Fusion der viralen mit der endosomalen Membran auslösen und dadurch das RNP ins Zytoplasma freisetzen. [ 34 ] Bei Viren spielt RV G eine entscheidende Rolle bei der Virusaufnahme, höchstwahrscheinlich durch Wechselwirkungen mit mutmaßlichen zellulären Rezeptoren, die eine schnelle Aufnahme ermöglichen. In diesem Zusammenhang wurde gezeigt, dass die Pathogenität von an Gewebekulturen adaptierten RV-Stämmen (z. B. ERA, HEP und CVS-11) mit dem Vorhandensein einer Determinante an der antigenen Stelle III des G-Proteins korreliert. [ 35 ] Eine Arg → Gln-Mutation an Position 333 dieser antigenen Stelle des ERA-G-Proteins führte zu einer siebenfachen Verzögerung der Internalisierung der Gln333-RV-Variante im Vergleich zur Wildtyp-Variante. Die Asn194→Lys194-Mutation in RV G, die das Wiederauftreten des pathogenen Phänotyps erklärt, war mit einer signifikanten Verkürzung der Internalisierungszeit verbunden.[ 36 ] Darüber hinaus zeigten Experimente mit chimären RVs, dass die für die Internalisierung von RV-Virionen erforderliche Zeit signifikant erhöht und die Pathogenität stark reduziert war, nachdem das G-Gen des hochpathogenen SB-RV-Stamms, der aus einem cDNA-Klon des aus Silber gewonnenen, mit Fledermäusen assoziierten Stamms RV-18 stammte,[ 37 ] durch das des hochattenuierten SN-Stamms ersetzt wurde, der aus einem cDNA-Klon des SAD B19-RV-Impfstoffstamms isoliert wurde.[ 38 ] Zusammen stützen diese Daten die Annahme, dass die Kinetik der Virusaufnahme, die eine Funktion von RV G ist, ein wichtiger Faktor ist, der die Pathogenität von RV bestimmt.
- Virale Elemente, die an der Verbreitung und Übertragung von Viren beteiligt sind
Eine einzigartige Eigenschaft des Tollwutvirus ist seine Fähigkeit, sich von Zelle zu Zelle auszubreiten. Die Beobachtung, dass die Gln333 ERA-Variante in vitro ihre pH-abhängige Zell-Zell-Fusionsaktivität verliert [ 39 ] und eine stark verringerte Fähigkeit zur Ausbreitung von Zelle zu Zelle zeigt [ 40 ], legt nahe, dass RV G ebenfalls eine Schlüsselrolle bei der Zell-zu-Zell-Ausbreitung und somit der Virusübertragung spielt, wahrscheinlich durch seine fusiogene Aktivität. Diese Möglichkeit wird zusätzlich durch den Befund gestützt, dass die Ausbreitungsrate des pathogenen RV-Revertanten SPBNGAK fast doppelt so hoch ist wie die der nicht-pathogenen SPBNGA-Variante. Interessanterweise verursachte die Asn194 → Lys194-Mutation in G SPBNGAK eine Verschiebung des pH-Schwellenwerts für die Membranfusion zu einem höheren pH-Wert, was die Hypothese stützt, dass ein höherer pH-Schwellenwert für die Membranfusion mit einer verstärkten Virusausbreitung verbunden ist. [ 41 ]
Studien zu transneuronalen Indikatoren einer RV-Infektion bei Ratten [ 42 ] und Rhesusaffen [ 43 ] haben gezeigt, dass das Tollwutvirus in Axonen ausschließlich retrograd wandert. Obwohl verschiedene RV-Proteine an neuronalen Transportmechanismen beteiligt sind, scheint RV G eine vorherrschende Rolle bei der transneuronalen Ausbreitung der RV-Infektion zu spielen. Während beispielsweise eine periphere Infektion mit dem Virus der equinen infektiösen Anämie (EIAV), das mit RV G pseudotypisiert ist, zu einer Virusübertragung ins Rückenmark führt, gelang es demselben EIAV, das mit dem Virus der vesikulären Stomatitis G pseudotypisiert ist, nicht, in das Nervensystem einzudringen. [ 44 ] Darüber hinaus wurde festgestellt, dass die virale Ausbreitung des ERA G Arg 333 → Gln 333-Mutanten im ZNS im Vergleich zum Wildtyp-Mutanten stark reduziert ist, was erneut auf eine Funktion von intaktem RV G bei der transsynaptischen Ausbreitung hindeutet. Der überzeugendste Beweis für die wichtige Rolle von RV G im transsynaptischen Transport stammt jedoch aus der intrakraniellen Infektion von Mäusen mit einem rekombinanten G-defizienten RV-Virus. Dabei blieb die Infektion auf die Neuronen an der Inokulationsstelle beschränkt, ohne dass eine Ausbreitung auf sekundäre Neuronen beobachtet wurde.[ 45 ] Es ist jedoch wahrscheinlich, dass neben RV G auch RV M eine Rolle bei der Virusverbreitung und damit beim transsynaptischen Transport spielt. In diesem Zusammenhang wurde gezeigt, dass die Verbreitung der chimären SN-BMBG RV-Variante, die sowohl M als auch G aus dem hochpathogenen SB enthält, signifikant höher war als die Verbreitung der chimären SN-BG- oder SN-BM-Variante, die G bzw. M aus SB enthalten. Dies legt nahe, dass die optimale Interaktion von M mit G eine wichtige Rolle bei der Virusverbreitung von Zelle zu Zelle spielen könnte. [ 46 ] Da RV M die Virusknospung unterstützt, [ 47 ] ist es wahrscheinlich, dass die effizientere Ausbreitung der chimären RV SN-BMBG-Variante auf eine optimale Virusknospung an der postsynaptischen Membran zurückzuführen ist.
Jüngste Studien haben gezeigt, dass die Interaktion zwischen RV P und der leichten Kette von Dynein das RV RNP mit dem Transportsystem der Wirtszelle verbindet und dadurch den retrograden axonalen Transport des Virus erleichtert.[ 48 ],[ 49 ] Eine periphere Infektion erwachsener Mäuse zeigte jedoch, dass die Deletion der LC8-Bindungsdomäne von RV P den Eintritt des Virus in das ZNS nicht verhindert, was darauf hindeutet, dass das RV-Protein nicht direkt an der retrograden axonalen Ausbreitung von RV beteiligt ist.[ 50 ]
- Virale Elemente, die die Virusreplikation steuern
Anders als bei vielen anderen Viren, wie etwa dem Influenzavirus, ist die Pathogenität von RV umgekehrt proportional zur Rate der viralen RNA-Synthese und der Produktion infektiöser Viruspartikel. Ein Vergleich der von verschiedenen chimären Viren produzierten Mengen viraler mRNA und genomischer RNA legt nahe, dass die Transkription und Replikation viraler RNA durch mehrere Faktoren reguliert wird, darunter RV M, das als trans-agierender Faktor identifiziert wurde und den Wechsel von anfänglich hohen mRNA-Synthesewerten zur Synthese genomischer RNA vermittelt.[ 51 ] Darüber hinaus ist M aller Rhabdoviren in der Lage, die virale Genexpression durch Bindung an das RNP zu unterbinden, wodurch eine stark verdichtete, rückgratartige Struktur entsteht, die die RNA-Synthese nicht mehr unterstützen kann.
Um weitere virale Elemente zu identifizieren, die die Pathogenität durch Regulierung der Virusreplikation kontrollieren, wurden die 5'-terminalen Sequenzen des hochpathogenen SB-Stammes schrittweise durch Sequenzen des hochattenuierten SN-Impfstoffstamms ersetzt. Das Ergebnis waren die rekombinanten Viren SB2 (terminale Sequenz [TS] + L), SB3 (TS + L + Pseudogen [Ψ]), SB4 (TS + L + Ψ + G) und SB5 (TS + L + Ψ + G + M). Intramuskuläre Infektionen mit den parentalen SB- und SN-Viren sowie den chimären RVs SB2, SB3, SB4 und SB5 führten bei SB-infizierten Mäusen zu den höchsten Mortalitätsraten, während bei SN-infizierten Mäusen weder Morbidität noch Mortalität auftraten. Der Ersatz von TS, L und SB durch die entsprechenden Elemente von SN führte zu einer moderaten Reduktion von Morbidität und Mortalität, und ein zusätzlicher G- oder G-plus-M-Austausch reduzierte die virale Pathogenität stark oder verhinderte sie vollständig.
Die phänotypische Charakterisierung dieser Wildtyp- und chimären RVs in Gewebekulturen ergab, dass die Pathogenität eines bestimmten RV umgekehrt proportional zu seiner Fähigkeit zur Replikation in neuronalen Zellen ist. Obwohl SB sich in fast 1000-mal niedrigeren Konzentrationen als SN replizierte und der Ersatz von TS, L und in SB durch SN-Konzentrationen wenig Einfluss auf die virale Wachstumskinetik hatte, führte der zusätzliche Ersatz von G oder G plus M von SB durch die entsprechenden SN-Gene zu einer Erhöhung der Virusproduktion um 1 log. Dies legt nahe, dass die Replikationskinetik der viralen RNA sowie die Viruspartikelproduktion weitgehend durch das RV-G-Protein gesteuert werden. Diese Schlussfolgerung wird durch Daten gestützt, die mit RV-G-Varianten gewonnen wurden, die sich in ihren G-Proteinen in einer Aminosäure unterscheiden. Die pathogene Tollwutvirusvariante SPBNGAK 194 erzeugte in NA-Zellen einen Virustiter, der 1 log niedriger war als der der nicht-pathogenen Variante SPBNGAN 194, und Echtzeit-PCR-Analysen zeigten, dass die Transkriptions- und Replikationsraten viraler RNA in SPBNGAK-infizierten NA-Zellen fünf- bis zehnmal höher waren als in SPBNGAK-infizierten NA-Zellen.[ 52 ] Weitere Hinweise auf eine inverse Korrelation zwischen Pathogenität und der Syntheserate viraler RNA sowie der Produktion viraler Partikel lieferten Mäuse, die mit chimären rekombinanten Viren infiziert waren, in denen die G- und M-Gene des abgeschwächten SN-Stamms durch die des hochpathogenen SB-Stamms ersetzt waren. Diese Experimente ergaben eine signifikant erhöhte Pathogenität des elterlichen SN-Stamms mit RV G gegenüber dem pathogenen SB-Stamm. Die Pathogenität wurde weiter erhöht, als sowohl G als auch M von SB in SN eingeführt wurden.
Der Austausch von G oder M oder beiden in SN durch die entsprechenden Gene aus SB war mit einer signifikanten Verringerung der Viruspartikelproduktion sowie der viralen RNA-Synthese verbunden. Diese Daten deuten darauf hin, dass sowohl G als auch M eine wichtige Rolle in der RV-Pathogenese spielen, indem sie die virale Replikation regulieren. Die Feststellung, dass der Austausch von G oder G plus M in SN durch G oder G plus M aus SB zu einer moderaten bis starken Verringerung der viralen RNA-Transkription bzw. -Replikation führt, während der Austausch von M allein in SN durch M aus SB zu einer starken Zunahme der viralen RNA-Transkription und -Replikation führt, weist darauf hin, dass RV G auch eine wichtige regulatorische Funktion in der viralen RNA-Transkription/-Replikation hat, entweder allein oder durch Interaktion mit dem M-Protein. Der Mechanismus, durch den das RV-G-Gen die virale RNA-Synthese steuert, ist unbekannt. Bestimmte Nukleotidsequenzen innerhalb der RV-G-Gene, wie beispielsweise die Codons für Arg333 und Lys194, wurden als Zielmoleküle für zelluläre miRNAs identifiziert. Es wurde gezeigt, dass die Zielerkennung durch zelluläre miRNAs zu einer positiven oder negativen Regulation der viralen Replikation führen kann. [ 53 ] Arg 333 → Glu 333 oder Lys 194 → Ser 194 Substitutionen innerhalb der RV G-Gensequenz führen zur Abschaffung von miRNA-Zielsequenzen, was wiederum mit einer signifikanten Erhöhung der viralen RNA-Syntheserate einhergeht [Faber M, Thomas Jefferson University, PA, USA, unveröffentlichte Daten], was darauf hindeutet, dass zelluläre miRNAs des Wirts auch eine wichtige Rolle bei der Regulation der RV-Replikation spielen, wie dies für andere RNA-Viren, einschließlich des Vesikulären Stomatitis-Virus und HCV, gezeigt wurde. [ 54 ], [ 55 ]
Die Regulierung der Virusreplikation scheint einer der wichtigsten Mechanismen der RV-Pathogenese zu sein. Um der Immunantwort zu entgehen und die Integrität des neuronalen Netzwerks zu erhalten, können pathogene RV-Stämme, nicht jedoch abgeschwächte Stämme, ihre Wachstumsrate regulieren. Eine geringere Replikationsrate kommt pathogenen RV-Stämmen wahrscheinlich zugute, da sie die neuronale Struktur, die diese Viren nutzen, um das ZNS zu erreichen, erhält. Eine weitere Erklärung für die geringere Replikationsrate pathogener RV ist, dass das Virus, um einer frühzeitigen Erkennung durch das Immunsystem des Wirtes zu entgehen, die Expression seiner Antigene auf ein Minimum beschränkt.
Zusammenhang zwischen RV G-Expression, Apoptose und Pathogenität
Es ist bekannt, dass Straßentollwutvirusstämme, die deutlich pathogener sind als an Gewebekulturen adaptierte Stämme, sehr begrenzte Mengen an G exprimieren und erst spät im Infektionszyklus Apoptose induzieren. Dies legt nahe, dass die Pathogenität eines bestimmten Virusstamms umgekehrt proportional zur RV-G-Expression und der Fähigkeit zur Apoptoseinduktion ist.[ 56 ] Direkte Hinweise auf einen Zusammenhang zwischen der Menge der G-Expression und dem Ausmaß der Apoptose wurden mit dem rekombinanten RV SPBNGA-GA erhalten, der zwei identische G-Gene trug und RV-G überexprimierte.[ 57 ] Morphologische Untersuchungen an neuronalen Kulturen, die mit diesem rekombinanten RV infiziert wurden, zeigten, dass der Zelltod parallel zur RV-G-Überexpression signifikant zunahm und dass Apoptose der wichtigste Mechanismus beim RV-G-vermittelten Zelltod ist. Insbesondere die verringerte F-Aktin-Färbung nach einer SPBNGA-GA-Infektion ist konsistent mit einer durch Apoptose induzierten Depolymerisation von Aktinfilamenten. Außerdem war die Zahl der TUNEL-positiven Kerne in SPBNGA-GA-infizierten Neuronen im Vergleich zu nicht infizierten und SPBNGA-infizierten Neuronen signifikant erhöht. Der Mechanismus, durch den das RV G-Gen den apoptotischen Signalprozess vermittelt, ist jedoch weitgehend unbekannt. Es wurde vermutet, dass die RV G-Expression über einem bestimmten Schwellenwert die Zellmembran schwer zerstört. Es ist sehr wahrscheinlich, dass apoptotische Zellen im ZNS nicht schnell beseitigt werden und daher eine sekundäre Nekrose erleiden. [ 58 ] Andererseits kann eine RV-Infektion und insbesondere eine Überexpression des RV G-Proteins zu Pyroptose führen, einem der Apoptose ähnlichen Zelltodweg, der aber, anders als Apoptose, die Aktivierung von Caspase 1 beinhaltet und dadurch zur Nekrose führt. [ 59 ] Das Ausmaß der durch eine RV-Infektion induzierten Nekrose oder Pyroptose spielt wahrscheinlich eine entscheidende Rolle bei der Induktion der antiviralen Immunität. Während apoptotische Zellen ihre Membranintegrität bewahren und die angeborene Immunantwort nicht stimulieren, werden nekrotische Zellen permeabilisiert und sezernieren endogene Adjuvantien, die eine robuste angeborene Immunantwort auslösen können. [ 60 ]
Da der Grad der Apoptose/Nekrose mit der RV-Immunogenität korreliert, wurde vermutet, dass die immunstimulierende Wirkung apoptotischer/nekrotischer Zellen höchstwahrscheinlich zur Entstehung einer schützenden Immunantwort beiträgt. Daher ist die Regulierung der RV-G-Expression sehr wahrscheinlich ein wichtiger Faktor in der Tollwutpathogenese, da sie das Überleben und die Verbreitung pathogener RV-Varianten im Nervensystem ermöglicht, ohne offensichtliche neuronale Schäden zu verursachen und eine schützende Immunantwort auszulösen, die eine Infektion verhindern würde.
Die RV-G-Expression kann auf der Ebene der RNA-Synthese, der posttranslationalen Ebene oder auf beiden reguliert werden. Die von verschiedenen chimären RV-Varianten exprimierten RV-G-Konzentrationen spiegeln sich nachweislich in der viralen RNA-Syntheserate wider. Dies legt nahe, dass die unterschiedliche Regulation der RV-G-Expression durch diese Varianten auf Variationen in der viralen mRNA-Transkriptionsrate zurückzuführen ist. Wie bei den viralen RNA-Transkriptionsraten korreliert auch die von diesen Varianten exprimierte RV-G-Menge umgekehrt mit der viralen Pathogenität. Andererseits führte die Infektion primärer neuronaler Kulturen mit der weniger pathogenen RV-Variante CVS-B2c zu viermal höheren G-Protein-Konzentrationen als eine Infektion mit der hochpathogenen Variante CVS-N2c, obwohl bei beiden Infektionen vergleichbare G-mRNA-Konzentrationen synthetisiert wurden. Pulse-Chase-Experimente zeigten, dass die höheren G-Protein-Konzentrationen in CVS-B2c-infizierten Neuronen größtenteils auf eine geringere Abbaurate des CVS-B2c-G-Proteins im Vergleich zum CVS-N2c-G-Protein zurückzuführen waren. Der Mechanismus, der zum schnelleren proteolytischen Abbau des CVS-N2c-G-Proteins führt, muss jedoch noch geklärt werden.
Symptome Tollwut
Die Inkubationszeit für Tollwut beträgt durchschnittlich 30-90 Tage. Bei einer massiven Infektion durch große Wunden an Kopf und Gesicht kann sie auf 12 Tage verkürzt werden. In seltenen Fällen kann die Inkubationszeit 1 Jahr oder länger dauern.
Es gibt einen streng sequentiellen Wechsel von drei Krankheitsperioden: Prodromalphase, Erregungsphase, Lähmungsphase.
Die Prodromalphase beginnt mit dem Auftreten von drückenden oder ziehenden Schmerzen an der Bissstelle sowie Schmerzen entlang der Nerven. Im Bereich der Narbe kann es zu Brennen, Juckreiz, manchmal Rötung und Schwellung kommen. Der Patient verspürt allgemeines Unwohlsein, Kopfschmerzen und Übelkeit. Es treten Erbrechen, ein Anstieg der Körpertemperatur auf 37,5–38 ° C und Symptome einer fortschreitenden psychischen Störung auf: erhöhte Reflexerregbarkeit, ein unerklärliches Gefühl von Angst, Furcht, Melancholie. Oft ist der Patient depressiv, gehemmt, zurückgezogen, verweigert das Essen, schläft schlecht, klagt über düstere Gedanken und beängstigende Träume. Die Prodromalphase dauert 2–3 Tage, manchmal bis zu 7 Tage. Am Ende dieser Phase können Angstanfälle mit kurzzeitigen Atembeschwerden, einem Engegefühl in der Brust, begleitet von Tachykardie und erhöhter Atemfrequenz, auftreten.
Die Erregungsphase ist durch das Auftreten von Hydrophobie gekennzeichnet: Beim Versuch zu trinken und dann beim Anblick von Wasser oder der Erinnerung daran verspürt der Patient einen krampfartigen Krampf des Rachens und Kehlkopfes, bei dem er den Wasserbecher schreiend wegschüttet, zitternde Hände nach vorne wirft, Kopf und Körper zurückwirft. Der Hals ist gestreckt, eine schmerzhafte Grimasse verzerrt das Gesicht, das durch einen Krampf der Atemmuskulatur bläulich wird. Die Augen treten hervor, drücken Angst aus, flehen um Hilfe, die Pupillen sind erweitert, das Einatmen ist schwierig. Auf dem Höhepunkt des Anfalls sind Herz- und Atemstillstand möglich. Der Anfall dauert einige Sekunden, danach scheint sich der Zustand des Patienten zu bessern. Anschließend können Anfälle von Krämpfen der Kehlkopf- und Rachenmuskulatur sogar durch Luftbewegung (Aerophobie), helles Licht (Photophobie) oder ein lautes Wort (Akustikophobie) auftreten. Die Anfälle gehen mit psychomotorischer Unruhe einher, bei der sich der Patient wie ein „Verrückter“ verhält. Das Bewusstsein ist während des Anfalls getrübt, klärt sich aber in der interiktalen Phase. Während der Unruhe kommt es aufgrund des erhöhten Tonus des sympathischen Nervensystems zu einem starken Anstieg des Speichelflusses (Sialorrhoe) mit der Unfähigkeit, Speichel aufgrund von Krämpfen der Rachenmuskulatur zu schlucken. Der Patient spritzt Speichel. Bei manchen Patienten können Anzeichen von Meningismus und sogar Opisthotonus auftreten, und Krämpfe sind häufig. In diesem Fall kann sich die Zerebrospinalflüssigkeit nicht verändern, bei manchen Patienten kann jedoch die Proteinkonzentration ansteigen und die Zellzahl aufgrund von Lymphozyten zunehmen.
Ohne angemessene Behandlung verstärken sich die Anzeichen einer Dehydration, die Gesichtszüge werden schärfer und das Körpergewicht nimmt ab. Die Körpertemperatur steigt auf hohe Werte. Krämpfe sind möglich. Die Dauer der Erregungsphase beträgt etwa 2–3 Tage, selten 4–5 Tage. Ein tödlicher Ausgang tritt in der Regel während eines Anfalls ein. Selten überlebt der Patient das dritte Stadium der Krankheit.
Während der Lähmung beruhigt sich der Patient. Die Anfälle von Tollwut hören auf, der Patient kann trinken und Nahrung schlucken, sein Bewusstsein ist klar. Trotz des scheinbaren Wohlbefindens treten jedoch bald Lethargie, Apathie, Depression, Lähmungen der Gliedmaßen, Beckenbeschwerden und Lähmungen der Hirnnerven auf. Die Körpertemperatur steigt auf 42–43 °C, der Blutdruck sinkt, und am Ende des ersten Tages tritt der Tod durch Lähmung des Herz-Kreislauf- und Atmungszentrums ein.
Im peripheren Blut werden neutrophile Leukozytose, erhöhte Hämoglobin-, Erythrozyten- und Hämatokritwerte beobachtet.
Was bedrückt dich?
Formen
Klinisch unterscheidet man typische und atypische Formen. Zu den atypischen Formen zählen alle Fälle ohne Erregung und Hydrophobie. Zu den atypischen Formen zählen bulbäre, zerebelläre, meningoenzephalische usw.
Diagnose Tollwut
Der Nachweis von Tollwutantigen, Antikörpern, viraler RNA oder Virusisolierung ermöglicht die Diagnose von Tollwut. Da bei einem Tollwutpatienten jeder einzelne Test negativ ausfallen kann, sind manchmal serielle Serumproben zum Nachweis von Tollwutantikörpern, Speichelproben zur Viruskultur und eine Hautbiopsie zur direkten Immunfluoreszenzuntersuchung auf virales Antigen notwendig, insbesondere bei starkem Tollwutverdacht.
Eine der schnellsten Methoden zur Diagnose von Tollwut beim Menschen vor dem Tod ist die Durchführung eines direkten Immunfluoreszenztests an einer Hautbiopsie im Nackenbereich zum Nachweis von Tollwutantigen. Der direkte Immunfluoreszenztest ist die sensitivste und spezifischste Methode zum Nachweis von Tollwutantigen in Haut und anderen frischen Geweben (z. B. Hirnbiopsie), obwohl die Ergebnisse im Frühstadium der Erkrankung gelegentlich negativ sein können. Steht kein frisches Gewebe zur Verfügung, kann die enzymatische Verdauung fixierter Gewebe die Reaktivität des Immunfluoreszenztests erhöhen; die Sensitivität kann jedoch inakzeptabel niedrig sein.
Die Diagnose kann auch gestellt werden, wenn das Virus nach Inokulation von Neuroblastomzellen oder Labornagern aus Speichel isoliert wird; dies ist in der Regel in den ersten zwei bis drei Krankheitswochen am effektivsten. Der Nachweis von Tollwutvirus-neutralisierenden Antikörpern im Serum ungeimpfter Personen, üblicherweise mittels Rapid Fluorescent Focus Inhibition Test (RFFIT), ist ebenfalls diagnostisch. Das Vorhandensein von Antikörpern im Liquor bestätigt die Diagnose, sie können jedoch zwei bis drei Tage später als Serumantikörper auftreten und sind daher in den frühen Stadien der Erkrankung weniger hilfreich. Während die serologische Reaktion nach der Impfung im Allgemeinen nicht von der durch die Krankheit induzierten serologischen Reaktion zu unterscheiden ist, führt die Impfung in der Regel nicht zur Bildung von Antikörpern im Liquor.
Nur sieben Fälle einer Tollwut-Genesung in den letzten 25 Jahren wurden gut dokumentiert. Obwohl bei keinem der Patienten das Tollwutvirus isoliert werden konnte, stützten hohe Titer von Tollwut-neutralisierenden Antikörpern in Serumproben und das Vorhandensein neutralisierender Antikörper in der Zerebrospinalflüssigkeit die Diagnose deutlich.
Was muss untersucht werden?
Welche Tests werden benötigt?
Differenzialdiagnose
Die Diagnose der menschlichen Tollwut wird üblicherweise anhand epidemiologischer und klinischer Daten gestellt und im Labor bestätigt. Die Diagnose ist eindeutig, wenn in der Anamnese Tierbisse vorliegen und das gesamte Spektrum der Symptome und Anzeichen aufgetreten ist. Andernfalls ist eine sorgfältige, aber schnelle Bewertung der epidemiologischen und klinischen Merkmale weniger typischer Fälle erforderlich, bevor spezifische Labortests durchgeführt werden. Jeder Patient mit neurologischen Anzeichen oder Symptomen oder einer ungeklärten Enzephalitis sollte nach einem möglichen Kontakt mit Tieren in Tollwut-Endemiegebieten innerhalb oder außerhalb des Wohnsitzlandes befragt werden. Der fehlende Verdacht auf Tollwut bei mehreren Todesfällen in den USA könnte auf eine unzureichende sorgfältige Anamnese zurückzuführen sein.
Zu Beginn der Erkrankung kann Tollwut viele infektiöse und nichtinfektiöse Krankheiten imitieren. Viele andere Enzephalitiden, beispielsweise durch Herpesviren und Arboviren, ähneln Tollwut. Auch andere Infektionskrankheiten wie Tetanus, zerebrale Malaria, Rickettsiose und Typhus können Tollwut imitieren. Zu den paralytischen Infektionskrankheiten, die mit Tollwut verwechselt werden können, gehören Poliomyelitis, Botulismus und Herpes-simian-B-Enzephalitis.
Zu den nichtinfektiösen Erkrankungen, die mit Tollwut verwechselt werden können, gehören eine Reihe neurologischer Syndrome, insbesondere die akute entzündliche Polyneuropathie (Guillain-Barré-Syndrom), sowie die allergische Enzephalomyelitis nach einer Tollwutimpfung des Nervengewebes, Vergiftungen oder Drogenintoxikationen, Alkoholentzug, akute Porphyrie und Tollwuthysterie. Das Guillain-Barré-Syndrom kann mit paralytischer Tollwut verwechselt werden und umgekehrt.
Wen kann ich kontaktieren?
Behandlung Tollwut
Eine Behandlung gegen Tollwut wurde bisher nicht entwickelt. Die Gabe hoher Dosen spezifischer Tollwut-Immunglobuline und Leukozyten-Interferone ist wirkungslos. Eine symptomatische Behandlung soll das Leiden des Patienten lindern. Zu diesem Zweck wird der Patient in einer separaten Station oder Box untergebracht und ein Schutzregime geschaffen, das den Einfluss der äußeren Umgebung (reduzierter Lärm, helles Licht, Luftstrom) begrenzt. Um die Erregbarkeit des Zentralnervensystems zu verringern, werden Schlafmittel, Antikonvulsiva und Schmerzmittel verschrieben. Der Wasserhaushalt wird normalisiert.
Im paralytischen Stadium werden Medikamente verschrieben, die die Aktivität des Herz-Kreislauf- und Atmungssystems stimulieren. Es wird empfohlen, hyperbare Sauerstoffversorgung, zerebrale Hypothermie und kontrollierte künstliche Beatmung mit vollständiger Kurarisierung des Patienten anzuwenden. Alle Behandlungsmethoden sind jedoch praktisch wirkungslos. Im besten Fall ist es möglich, das Leben des Patienten um mehrere Monate zu verlängern. Ein ungünstiger Ausgang wird durch die Schwere der Schädigung des Hirnstamms mit der Zerstörung lebenswichtiger Zentren vorbestimmt.
Verhütung
Die Entwicklung des ersten Tollwutimpfstoffs durch Pasteur im Jahr 1885 läutete eine Ära deutlich effektiverer Tollwutbekämpfung ein. Heute ist die Krankheit trotz einer fast 100%igen Sterblichkeitsrate beim Menschen durch Tollwut durch eine Impfung vor und/oder nach der Exposition vollständig vermeidbar. Während Pasteur und seine Kollegen die Impfung von Privathunden in Paris einführten, fand die erste Massenimpfung von Hunden Anfang der 1920er Jahre in Japan statt und markierte damit das erste große nationale Tollwutbekämpfungsprogramm. Die orale Impfung von Wildtieren, die erstmals in den 1970er Jahren entwickelt wurde, hat sich seitdem wiederholt als wirksam bei der Bekämpfung der Krankheit bei wichtigen terrestrischen Wirten wie Füchsen, Waschbären und Stinktieren erwiesen. [ 68 ] Eine anhaltende Tollwutimpfung von Reservoirtierpopulationen mit einer Durchimpfungsrate von 70 % oder mehr wird RABV schließlich aus Reservoirarten eliminieren und die Ausbreitung des Virus auf Nebenwirte verhindern. [ 69 ]
Phylogenetische Daten deuten darauf hin, dass Lyssaviren Fledermäuse lange vor Landsäugetieren infizierten, und die meisten Lyssaviren, einschließlich RABV, zirkulieren noch immer weltweit in verschiedenen Fledermausarten.[ 70 ] Wirksame Methoden zur Verhinderung der Übertragung von RABV unter Fledermäusen sind jedoch nach wie vor schwer fassbar, sodass eine vollständige Ausrottung der Tollwut derzeit nicht möglich ist. Selbst nach Kontakt mit RABV durch den Biss eines tollwutinfizierten Säugetiers kann eine sichere und wirksame Postexpositionsprophylaxe (PEP, einschließlich Wundreinigung, Tollwutimmunglobulin und Tollwutimpfung) Menschen vor einer Tollwutinfektion schützen, wenn die Behandlung umgehend und gemäß den Empfehlungen der Weltgesundheitsorganisation (WHO) erfolgt.
Diese beiden Methoden zur Verhinderung menschlicher Todesfälle – die eine basiert auf der Impfung exponierter Personen, die andere auf der Impfung einer ausreichenden Anzahl von Hunden, um den Übertragungszyklus an der Quelle zu unterbrechen – sind die Bausteine eines „One-Health“-Ansatzes zur Prävention und Bekämpfung der Tollwut bei Hunden. Diese beiden unterschiedlichen Methoden zur Verhinderung menschlicher Todesfälle wurden als separate Alternativen betrachtet: Strategie A, basierend auf der Bereitstellung von PEP für Menschen, und Strategie B, basierend auf der Impfung von Hunden; oder als Komponenten einer kombinierten Strategie A + B in einer Analyse der voraussichtlichen Kosten der alternativen Strategien.[ 71 ]
Länder wie Thailand konnten durch den Einsatz von PEP enorme Erfolge bei der Verhinderung menschlicher Todesfälle erzielen, mussten jedoch auch feststellen, dass die Nachfrage nach PEP allein zunimmt und die damit verbundenen Kosten steigen. [ 72 ] Im Jahr 2003 benötigten beispielsweise im Vergleich zur Situation im Jahr 1991 viermal so viele Menschen (über 400.000) PEP. Aktuelle Daten zeigen, dass die Volksrepublik China, die jährlich 15 Millionen Menschen nach möglichem Tollwutexposition impft, allein für PEP jährlich etwa 650 Millionen US-Dollar ausgibt. [ 73 ]
Ein wesentlich nachhaltigerer Ansatz besteht darin, die Ausbreitung der Infektion an der Quelle, im Tierbestand, zu verhindern und gleichzeitig den Zugang zu PEP für exponierte menschliche Patienten bei Bedarf zu verbessern. Wo politischer Wille und ausreichende Finanzierung zur Bekämpfung der Tollwut bei Hunden vorhanden sind, können Todesfälle vermieden werden, und dies ist auch bereits der Fall. Die flächendeckende Impfung von Hunden hat zur Eliminierung der Tollwut bei Hunden in mehreren Ländern geführt, darunter in Malaysia 1954, [ 74 ] Japan 1956, Taiwan 1961, Singapur und insbesondere in ganz Westeuropa (siehe Rupprecht et al., King et al. sowie Gongal und Wright). [ 75 ]
Использованная литература