Facharzt des Artikels

Neue Veröffentlichungen
Charcot-Marie-Tooth-Krankheit.
Zuletzt überprüft: 12.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
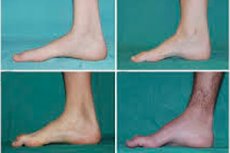
Peroneale Muskelatrophie, auch Charcot-Marie-Tooth-Syndrom oder -Krankheit genannt, ist eine Gruppe chronischer Erbkrankheiten mit Schädigungen der peripheren Nerven.
Laut ICD-10 lautet der Code für diese Krankheit im Abschnitt über Erkrankungen des Nervensystems G60.0 (hereditäre motorische und sensorische Neuropathie). Es ist auch in der Liste der seltenen Krankheiten enthalten.
Epidemiologie
Laut klinischer Statistik beträgt die Prävalenz aller Arten der Charcot-Marie-Tooth-Krankheit pro 100.000 Einwohner 19 Fälle (anderen Quellen zufolge ein Fall pro 2,5 bis 10.000 Einwohner).
CMT Typ 1 macht etwa zwei Drittel der Fälle aus (ein Fall pro 5.000 bis 7.000 Einwohner), und fast 70 % davon sind mit einer Duplikation des PMP22-Gens verbunden. Weltweit leiden mehr als 1,2 Millionen Menschen an dieser Erkrankung.
Die Inzidenz von CMT Typ 4 wird auf 1-5 Fälle pro 10.000 Kinder geschätzt. [ 1 ]
Ursachen Charcot-Marie-Tooth-Krankheit
Gemäß der Klassifikation der polyneuropathischen Syndrome handelt es sich bei der peronealen (fibulären) Muskelatrophie, der Charcot-Marie-Tooth-Neuralamyotrophie oder der Charcot-Marie-Tooth-Krankheit (abgekürzt CMT) um genetisch bedingte motorisch-sensorische Polyneuropathien. [ 2 ]
Das heißt, die Ursachen für sein Auftreten sind genetische Mutationen. Abhängig von der Art der genetischen Abweichungen werden die Haupttypen oder Arten dieses Syndroms unterschieden: demyelinisierend und axonal. Die erste Gruppe umfasst die Charcot-Marie-Tooth-Krankheit Typ 1 (CMT1), die aufgrund einer Duplikation des PMP22-Gens auf Chromosom 17 auftritt, das für das transmembranäre periphere Myelinprotein 22 kodiert. Infolgedessen kommt es zu einer segmentalen Demyelinisierung der Axonscheide (Nervenzellfortsätze) und einer Verringerung der Nervensignalleitungsgeschwindigkeit. Darüber hinaus können Mutationen in einigen anderen Genen vorliegen.
Die axonale Form ist die Charcot-Marie-Tooth-Krankheit Typ 2 (CMT2). Sie betrifft die Axone selbst und ist mit pathologischen Veränderungen im MFN2-Gen am Locus 1p36.22 verbunden. Dieses Gen kodiert das Membranprotein Mitofusin-2, das für die Mitochondrienfusion und die Bildung funktioneller mitochondrialer Netzwerke in peripheren Nervenzellen notwendig ist. Es gibt mehr als ein Dutzend Subtypen von CMT2 (mit Mutationen in bestimmten Genen).
Es ist zu beachten, dass derzeit mehr als hundert Gene identifiziert wurden, deren vererbte Schäden verschiedene Subtypen der Charcot-Marie-Tooth-Krankheit verursachen. Beispielsweise führen Mutationen im RAB7-Gen zu CMT Typ 2B; Veränderungen des SH3TC2-Gens (das für eines der Membranproteine der Schwann-Zellen kodiert) verursachen CMT Typ 4C, das sich im Kindesalter manifestiert und durch Demyelinisierung motorischer und sensorischer Neuronen gekennzeichnet ist (es gibt ein Dutzend und ein halbes Dutzend Formen von Typ 4 dieser Krankheit).
Eine seltene CMT vom Typ 3 (Dejerine-Sottas-Syndrom genannt) beginnt sich in der frühen Kindheit zu entwickeln und wird durch Mutationen in den Genen PMP22, MPZ, EGR2 und anderen verursacht.
Beim Auftreten der CMT Typ 5 im Alter von 5–12 Jahren kommt es neben einer motorischen Neuropathie (in Form einer spastischen Paraparese der unteren Extremitäten) auch zu einer Schädigung des Seh- und Hörnervs.
Typisch für den CMT Typ 6 sind Muskelschwäche und Optikusatrophie (mit Sehverlust) sowie Gleichgewichtsstörungen. Und beim Morbus Charcot-Marie-Tooth Typ 7 kommt es neben der motorisch-sensorischen Neuropathie auch zu einer Netzhauterkrankung in Form einer Retinitis pigmentosa.
Die X-chromosomale CMT oder Charcot-Marie-Tooth-Krankheit mit Tetraparese der Gliedmaßen (Bewegungsschwäche beider Arme und Beine) tritt häufiger bei Männern auf. Es handelt sich um eine demyelinisierende Form und man geht davon aus, dass sie auf eine Mutation im GJB1-Gen auf dem langen Arm des X-Chromosoms zurückzuführen ist. Dieses Gen kodiert Connexin 32, ein Transmembranprotein von Schwann-Zellen und Oligodendrozyten, das die Übertragung von Nervensignalen reguliert. [ 3 ]
Risikofaktoren
Der Hauptrisikofaktor für CMT ist eine familiäre Vorbelastung mit der Erkrankung, also bei nahen Verwandten.
Genetiker gehen davon aus, dass das Risiko, ein Kind mit dieser Krankheit zu bekommen, 25 % beträgt, wenn beide Elternteile Träger des autosomal-rezessiven Gens für die Charcot-Marie-Tooth-Krankheit sind. Das Risiko, dass das Kind Träger dieses Gens ist (aber keine Symptome zeigt), wird auf 50 % geschätzt.
Bei X-chromosomaler Vererbung (wenn sich das mutierte Gen auf dem X-Chromosom der Frau befindet) besteht ein 50-prozentiges Risiko, dass die Mutter das Gen an ihren Sohn vererbt, der an CMT erkrankt. Die Krankheit tritt möglicherweise nicht bei der Geburt eines Mädchens auf, aber die Söhne (Enkel) der Tochter können das defekte Gen erben, und die Krankheit entwickelt sich.
Pathogenese
Bei allen Formen der Charcot-Marie-Tooth-Krankheit liegt die Pathogenese in einer erblichen Anomalie der peripheren Nerven begründet: motorische (Bewegung) und sensorische (Empfindlichkeit).
Handelt es sich bei dem CMT-Typ um eine demyelinisierende Erkrankung, führt die Zerstörung oder der Defekt der Myelinscheide, die die Axone der peripheren Nerven schützt, zu einer Verlangsamung der Übertragung von Nervenimpulsen im peripheren Nervensystem – zwischen Gehirn, Muskeln und Sinnesorganen.
Beim axonalen Typ der Erkrankung sind die Axone selbst betroffen, was sich negativ auf die Stärke der Nervensignale auswirkt, die für eine vollständige Stimulation der Muskeln und Sinnesorgane nicht ausreicht.
Lesen Sie auch:
Wie wird das Charcot-Marie-Tooth-Syndrom übertragen? Die defekten Gene können autosomal-dominant oder autosomal-rezessiv vererbt werden.
Die häufigste Form, die autosomal-dominante Vererbung, tritt auf, wenn eine Kopie des mutierten Gens (bei einem Elternteil) vorhanden ist. Die Wahrscheinlichkeit, CMT an jedes Kind weiterzugeben, wird auf 50 % geschätzt. [ 4 ]
Bei der autosomal-rezessiven Vererbung sind für die Entstehung der Krankheit zwei Kopien des defekten Gens (eine von jedem Elternteil, der keine Krankheitsanzeichen zeigt) erforderlich.
In 40-50 % der Fälle kommt es zu einer autosomal-dominanten Demyelinisierung (CMT Typ 1), in 12-26 % zu einer axonalen CMT (Typ 2). In 10-15 % der Fälle wird ein X-chromosomaler Erbgang beobachtet. [ 5 ]
Symptome Charcot-Marie-Tooth-Krankheit
Typischerweise treten die ersten Anzeichen dieser Krankheit im Kindes- und Jugendalter auf und entwickeln sich allmählich im Laufe des Lebens, obwohl sich das Syndrom auch später bemerkbar machen kann. Die Kombination der Symptome ist variabel, und das Fortschreiten der Krankheit sowie ihr Schweregrad sind nicht vorhersehbar.
Typische Symptome im Anfangsstadium sind erhöhte allgemeine Müdigkeit, verminderter Tonus (Schwäche) der Fuß-, Knöchel- und Schienbeinmuskulatur sowie fehlende Reflexe. Dies erschwert die Fußbewegung und führt zu Dysbasie (Gangstörung) in Form eines Hochhebens der Beine, oft mit häufigem Stolpern und Stürzen. Anzeichen der Charcot-Marie-Tooth-Krankheit bei kleinen Kindern können ausgeprägte Ungeschicklichkeit und altersuntypische Gehschwierigkeiten in Verbindung mit einem beidseitigen Fallfuß sein. Ebenfalls charakteristisch sind Fußdeformitäten: Hohlfuß oder starke Plattfüße, gekrümmte (hammerförmige) Zehen.
Beim Zehengang vor dem Hintergrund einer Muskelhypotonie kann ein Neurologe vermuten, dass das Kind an CMT Typ 4 leidet, bei der Kinder im Jugendalter möglicherweise nicht mehr laufen können.
Mit fortschreitender Erkrankung breiten sich Muskelschwund und -schwäche auf die oberen Extremitäten aus, was die Feinmotorik und die Ausführung normaler Handbewegungen erschwert. Verminderte Tast- und Kälteempfindungen sowie Taubheitsgefühle in Füßen und Händen deuten auf eine Schädigung der Axone der sensorischen Nerven hin.
Bei der Charcot-Marie-Tooth-Krankheit Typ 3 und 6, die sich im Kindesalter manifestiert, werden sensorische Ataxie (Beeinträchtigung der Bewegungs- und Gleichgewichtskoordination), Muskelzuckungen und Zittern, Schäden am Gesichtsnerv, Sehnervenatrophie mit Nystagmus und Hörverlust beobachtet.
In späteren Stadien kann es zu unkontrollierbarem Zittern (Tremor) und häufigen Muskelkrämpfen kommen; Bewegungsprobleme können zur Entwicklung von Muskel-, Gelenk- und neuropathischen Schmerzen führen.
Komplikationen und Konsequenzen
Die Charcot-Marie-Tooth-Krankheit kann Komplikationen und Folgen haben wie:
- häufigere Verstauchungen und Knochenbrüche;
- Kontrakturen, die mit einer Verkürzung der periartikulären Muskeln und Sehnen einhergehen;
- Skoliose (Verkrümmung der Wirbelsäule);
- Atemprobleme – wenn die Nervenfasern, die die Zwerchfellmuskulatur innervieren, beschädigt sind:
- Verlust der Fähigkeit, sich selbstständig zu bewegen.
Diagnose Charcot-Marie-Tooth-Krankheit
Die Diagnose umfasst eine klinische Untersuchung, eine Anamnese (einschließlich Familienanamnese) sowie eine neurologische und systemische Untersuchung.
Es werden Tests durchgeführt, um den Bewegungsumfang, die Sensibilität und die Sehnenreflexe zu überprüfen. Die Nervenleitgeschwindigkeit kann mittels instrumenteller Diagnostik – Elektromyographie oder Elektroneuromyographie – beurteilt werden. Ultraschall oder MRT können ebenfalls erforderlich sein. [ 6 ]
Genetische oder DNA-Tests zum Nachweis der häufigsten genetischen Mutationen, die CMT verursachen, in einer Blutprobe sind nur begrenzt möglich, da DNA-Tests derzeit nicht für alle Arten der Krankheit verfügbar sind. Weitere Informationen finden Sie unter Genetische Tests
In manchen Fällen wird eine Biopsie des peripheren Nervs (meist des Nervus suralis) durchgeführt.
Differenzialdiagnose
Zu den Differentialdiagnosen gehören andere periphere Neuropathien, Muskeldystrophie Duchenne, myelopathische und myasthenische Syndrome, diabetische Neuropathie, Myelopathien bei multipler und amyotropher Lateralsklerose, Guillain-Barré-Syndrom, Traumata des Nervus peroneus und dessen Atrophie (auch bei Einklemmung zwischen den Bandscheiben der Lendenwirbelsäule), Schäden am Kleinhirn oder Thalamus sowie Nebenwirkungen einer Chemotherapie (bei Behandlung mit Zytostatika wie Vincristin oder Paclitaxel). [ 7 ]
Wen kann ich kontaktieren?
Behandlung Charcot-Marie-Tooth-Krankheit
Die Behandlung dieser Erbkrankheit umfasst heute Bewegungstherapie (zur Stärkung und Dehnung der Muskulatur), Beschäftigungstherapie (zur Unterstützung von Patienten mit Muskelschwäche in den Händen) und den Einsatz orthopädischer Hilfsmittel zur Erleichterung des Gehens. Bei Bedarf werden Schmerzmittel oder Antiepileptika verschrieben. [ 8 ]
Bei ausgeprägten Plattfüßen kann eine Osteotomie durchgeführt werden, bei Fersendeformationen ist eine chirurgische Korrektur – die Arthrodese – angezeigt. [ 9 ]
Sowohl die genetische Komponente der Krankheit als auch ihre Behandlungsmethoden werden derzeit erforscht. Der Einsatz von Stammzellen, bestimmten Hormonen, Lecithin oder Ascorbinsäure hat bisher keine positiven Ergebnisse gebracht.
Dank neuerer Forschung könnte es in naher Zukunft tatsächlich neue Behandlungsmöglichkeiten für die Charcot-Marie-Tooth-Krankheit geben. So entwickelt das französische Unternehmen Pharnext seit 2014 das Medikament PXT3003 zur Behandlung von CMT Typ 1 bei Erwachsenen, und seit Mitte 2019 laufen klinische Studien dazu. PXT3003 unterdrückt die erhöhte Expression des PMP22-Gens, verbessert die Myelinisierung peripherer Nerven und lindert neuromuskuläre Symptome.
Spezialisten des US-amerikanischen Medizinunternehmens Sarepta Therapeutics arbeiten an einer Gentherapie für die Charcot-Marie-Tooth-Krankheit Typ 1. Bei dieser Therapie kommt ein harmloses Adeno-assoziiertes Virus (AAV) der Gattung Dependovirus mit linearem einzelsträngigem DNA-Genom zum Einsatz, das das NTF3-Gen in den Körper überträgt. Es kodiert das Protein Neurotrophin-3 (NT-3), das für die Funktion der Schwann-Nervenzellen notwendig ist.
Helixmith wird bis Ende 2020 mit klinischen Studien der in Südkorea entwickelten Gentherapie Engensis (VM202) zur Behandlung von Muskelsymptomen bei CMT Typ 1 beginnen. [ 10 ]
Verhütung
Eine genetische Beratung zukünftiger Eltern kann einer CMT vorbeugen, insbesondere wenn die Krankheit in der Familie eines Paares vorkommt. Es wurden jedoch auch Fälle von de novo Punktgenmutationen festgestellt, bei denen die Krankheit in der Familienanamnese nicht vorkommt.
Während der Schwangerschaft kann die Wahrscheinlichkeit einer Charcot-Marie-Tooth-Krankheit beim zukünftigen Kind durch eine Chorionzottenbiopsie (von der 10. bis zur 13. Schwangerschaftswoche) sowie eine Analyse des Fruchtwassers (in der 15. bis 18. Woche) überprüft werden.
Prognose
Die Prognose der verschiedenen Formen der Charcot-Marie-Tooth-Krankheit hängt im Allgemeinen vom klinischen Schweregrad ab. In allen Fällen verläuft die Krankheit jedoch langsam. Viele Patienten weisen Behinderungen auf, die jedoch die Lebenserwartung nicht verkürzen.