Facharzt des Artikels

Neue Veröffentlichungen
Subakute nekrotisierende Leah-Enzephalomyopathie
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Die Krankheit wurde erstmals 1951 erwähnt. Bis heute wurden mehr als 120 Fälle beschrieben. Die Leigh-Krankheit (OMIM 256000) ist eine genetisch heterogene Erkrankung, die entweder nukleär (autosomal-rezessiv oder X-chromosomal) oder mitochondrial (seltener) vererbt werden kann.
 [
[ Ursachen des Leah-Syndroms
Die Erkrankung beruht auf einem Mangel an Enzymen, die für die Energieproduktion sorgen. Der Grund dafür ist vor allem eine Störung des Brenztraubensäurestoffwechsels und ein Defekt im Elektronentransport in der Atmungskette. Es kommt zu einem Mangel des Pyruvat-Dehydrogenase-Komplexes (α-E1-Untereinheit), der Pyruvat-Carboxylase, des Komplexes 1 (NAD-Coenzym-Q-Reduktase) und des Komplexes 4 (Cytochromoxidase) der Atmungskette.
Es wurde festgestellt, dass Defekte der Pyruvatcarboxylase, des Komplexes 1 (NAD-Coenzym Q-Reduktase) und des Komplexes 4 (Cytochromoxidase) der Atmungskette autosomal-rezessiv vererbt werden, Defekte des Pyruvatdehydrogenasekomplexes (a-E1-Untereinheit) werden X-chromosomal-rezessiv vererbt. Bei Punktmutationen der mtDNA, die die 6. Untereinheit der ATPase betreffen, ist eine mitochondriale Vererbung typisch. Am häufigsten tritt eine Miscens-Mutation auf, die mit dem Ersatz von Thymin durch Guanin oder Cytosin an Position 8993 der mtDNA verbunden ist. Seltener ist eine Mutation an Position 9176 der mtDNA. Aufgrund der Tatsache, dass die T8993G-Mutation der Hauptdefekt beim NARP-Syndrom ist, wurden Familien mit diesen beiden Krankheiten beschrieben. Bei Kindern wurde zudem eine Mutation in der mtDNA an Position 8344 beschrieben, die beim MERRF-Syndrom auftritt.
Es wird angenommen, dass sich bei Ansammlung mutierter mtDNA in den meisten Mitochondrien ein schwerer Verlauf des Leigh-Syndroms entwickelt. In der mitochondrialen Genese dieser Erkrankung findet sich mutierte mtDNA in 90 % aller Mitochondrien. Die Pathogenese ist mit einer Störung der Energieproduktion in Zellen und der Entwicklung einer Laktatazidose verbunden.
Symptome des Leah-Syndroms
Die ersten Anzeichen der Krankheit treten bereits im frühen Alter (1–3 Jahre) auf. Es sind jedoch auch Fälle bekannt, in denen die Krankheit bereits im Alter von 2 Wochen und 6–7 Jahren auftritt. Zunächst entwickeln sich unspezifische Störungen: verzögerte psychomotorische Entwicklung, verminderter Appetit, Erbrechen, Gewichtsdefizit. Anschließend verstärken sich die neurologischen Symptome: Muskelhypotonie oder -dystonie mit Übergang in Hypertonie, myoklonische oder tonisch-klonische Anfälle, Tremor der Extremitäten, Choreoathetose, Koordinationsstörungen, verminderte Sehnenreflexe, Lethargie, Schläfrigkeit. Die zerebrale Neurodegeneration verläuft progressiv. Die Symptome einer Pyramiden- und Extrapyramidalinsuffizienz nehmen zu, der Schluckakt ist beeinträchtigt. Häufig treten Veränderungen des Sehorgans auf, wie Ptosis, Ophthalmoplegie, Sehnervenatrophie und seltener eine Pigmentdegeneration der Netzhaut. Manchmal entwickelt sich eine hypertrophe Kardiomyopathie und es treten Tachypnoe-Episoden auf.
In seltenen Fällen verläuft die Krankheit als akute Enzephalopathie. Typischer ist ein chronischer oder subakuter Verlauf, der mehrere Jahre nach Ausbruch der Krankheit zum Tod führt. Bei einem schnellen Verlauf (mehrere Wochen) tritt der Tod durch eine Lähmung des Atemzentrums ein.
Diagnose des Leah-Syndroms
Eine biochemische Blutuntersuchung zeigt eine Laktatazidose aufgrund der Ansammlung von Milchsäure und Brenztraubensäure im Blut und der Zerebrospinalflüssigkeit sowie eines Anstiegs des Alaningehalts im Blut. Auch der Ketonkörperspiegel kann erhöht sein. Im Urin wird eine erhöhte Ausscheidung organischer Säuren festgestellt: Milchsäure, Fumarsäure usw. Der Carnitinspiegel im Blut und Gewebe sinkt häufig.
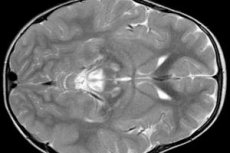
EEG-Ergebnisse zeigen fokale Anzeichen epileptischer Aktivität. MRT-Daten zeigen eine Vergrößerung der Hirnventrikel, bilaterale Hirnschäden und Verkalkung der Basalganglien (Nucleus caudatus, Putamen, Substantia nigra, Globus pallidus). Auch eine Atrophie der Großhirnhemisphären und der Hirnsubstanz kann nachgewiesen werden.
Die morphologische Untersuchung zeigt deutliche Veränderungen der Hirnsubstanz: symmetrische Nekroseherde, Demyelinisierung und schwammartige Degeneration des Gehirns, vor allem der Mittelabschnitte, der Brücke, der Basalganglien, des Thalamus und des Sehnervs. Das histologische Bild umfasst zystische Degeneration des Hirngewebes, astrozytische Gliose, neuronalen Tod und eine Zunahme der Mitochondrien in den Zellen. In der Skelettmuskulatur kommt es zu einer Ansammlung von Lipideinschlüssen, einer Abnahme der histochemischen Reaktion auf die Komplexe 1 und 4 der Atmungskette, einer subsarkolemmalen Ansammlung von Mitochondrien sowie abnormen Mitochondrien mit Desorganisation der Cristae. Das RRF-Phänomen wird oft nicht erkannt.
Wie zu prüfen?
Welche Tests werden benötigt?
Использованная литература