Facharzt des Artikels

Neue Veröffentlichungen
Neuroblastom bei Kindern: Ursachen, Diagnose, Behandlung
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
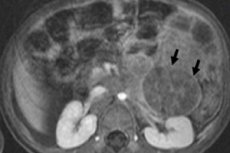
In der pädiatrischen Onkologie ist das Neuroblastom bei Kindern eine der häufigsten extrakraniellen Neoplasien. Dabei handelt es sich um einen embryonalen bösartigen Tumor der Neuroblasten der Neuralleiste, also der embryonalen (unreifen) Nervenzellen des sympathischen Nervensystems.
Epidemiologie
Laut Statistiken der International Neuroblastoma Risk Group (INRG) macht das Neuroblastom etwa 8 % aller onkologischen Erkrankungen bei Kindern weltweit aus und steht nach Leukämie und Hirntumoren an dritter Stelle.
Anderen Daten zufolge macht das Neuroblastom etwa 28 % aller Krebserkrankungen bei Säuglingen aus. Mehr als ein Drittel der Neuroblastomfälle werden bei Kindern unter einem Jahr diagnostiziert; das durchschnittliche Diagnosealter liegt bei 19–22 Monaten. Mehr als 90 % der diagnostizierten Fälle treten bei Kindern im Alter von zwei bis fünf Jahren auf (wobei Jungen überwiegen); die höchste Inzidenz wird im Alter von zwei bis drei Jahren beobachtet, und die Fälle bei Kindern über fünf Jahren machen weniger als 10 % aus.
Ursachen Neuroblastome
Bei der Untersuchung der Ursachen des Neuroblastoms kamen Forscher zu dem Schluss, dass dieser Tumor bei Kindern durch sporadische genetische Mutationen während der Embryogenese oder der frühen postnatalen Entwicklung entsteht. Die Ursache dieser Genveränderungen ist jedoch unbekannt, da kein Einfluss teratogener Umweltfaktoren nachgewiesen wurde.
Diese Tumoren können überall auftreten, unter anderem im Mediastinum, im Hals, im Bauchraum, in den Nebennieren, den Nieren, der Wirbelsäule und im Becken.
In seltenen Fällen kann ein Neuroblastom bei Säuglingen mit einer vererbten Mutation einhergehen. Insbesondere eine Mutation im Gen des Membranproteins CD246 auf Chromosom 2 – dem Enzym Tyrosinkinase ALK, das die interzelluläre Kommunikation gewährleistet und eine wichtige Rolle für die Funktion des Nervensystems spielt; im Gen des Proteins PHOX2B (auf Chromosom 4), das an der Reifung von Nervenzellen beteiligt ist.
Neuroblastome können auch mit der Neurofibromatose Typ 1 im Kindesalter,dem Beckwith-Wiedemann-Syndrom und einer hyperinsulinämischen Hypoglykämie (Nesidioblastose-Pankreatitis) in Zusammenhang stehen.
Risikofaktoren
Heute gelten Vererbung und das Vorhandensein dieses Tumors in der Familienanamnese sowie angeborene Anomalien im Zusammenhang mit Genmutationen während der intrauterinen Entwicklung als Risikofaktoren für die Entwicklung eines Neuroblastoms bei Kindern. Dies gilt insbesondere für Fälle, in denen mehrere Neoplasien in verschiedenen Organen auftreten.
Keiner der exogenen Faktoren, die das Risiko dieses Tumors erhöhen, wurde von den Forschern identifiziert.
Pathogenese
Der Entstehungsmechanismus von Neuroblastomen beruht auf Störungen der Differenzierung und Reifung von Neuralleistenzellen – bilateralen Zelllinien, die sich an den Rändern des Neuralrohrs aus der ektodermalen Keimschicht des menschlichen Embryos bilden. Diese Zellen wandern (bewegen sich) und differenzieren sich in viele Zelltypen: sensorische und autonome Neuronen, neuroendokrine Zellen und Zellen des Nebennierenmarks, Zellen des kraniofazialen Knorpels und der Knochen sowie Pigmentzellen.
Beim Neuroblastom reifen die migrierten Neuroblasten nicht, sondern wachsen und teilen sich weiter und bilden einen Tumor. Die Pathogenese seiner Entstehung ist mit folgenden Genmutationen verbunden:
- mit Duplikation eines Teils der Chromosomensequenz oder Duplikation von Abschnitten des LMO1-Gens auf Chromosom 11, das das RBTN1-Protein in den Neuralleistenzellen des Embryos kodiert;
- mit einer Veränderung der Kopienzahl des NBPF10-Gens auf Chromosom 1q21.1, das für das Protein DUF1220 kodiert, das die Proliferation menschlicher neuronaler Stammzellen steuert. Diese Erkrankungen führen entweder zu einer Duplikation dieses Chromosoms oder zu seiner Deletion – dem Fehlen eines Teils der DNA;
- mit Veränderungen im Tumorsuppressorgen ATRX (auf Chromosom Xq21.1);
- mit dem Vorhandensein zusätzlicher Kopien (Amplifikation) des N-Myc-Transkriptionsfaktor-Gens auf Chromosom 2, das für einen der Transkriptionsfaktoren (DNA-bindendes Protein) kodiert, der die Aktivität anderer Gene reguliert und die Proliferation von Vorläuferzellen während der Bildung von Proteinen für die Bildung von Geweben und Organen des Fötus steuert. Die Amplifikation dieses Gens macht es zu einem Onkogen, das eine Störung des Zellzyklus, erhöhte Zellproliferation und Tumorbildung hervorruft.
Symptome Neuroblastome
Die ersten Anzeichen eines Neuroblastoms sind unspezifisch und können Appetitlosigkeit (und Gewichtsverlust), Müdigkeit beim Füttern, Fieber und Gelenkschmerzen umfassen.
Die klinischen Symptome hängen von der Lage des Primärtumors und dem Vorhandensein von Metastasen ab (die in 60–73 % der Fälle auftreten).
Sehr häufig ist das primäre Neuroblastom im Nebennierenmark lokalisiert, das einen ähnlichen Ursprung wie Nervenzellen hat. Bei Kindern unter einem Jahr wird in 35-40 % der Fälle ein Nebennierenneuroblastom diagnostiziert. Zu den Symptomen gehören Bauchschmerzen, Fieber, Gewichtsverlust, Knochenschmerzen, Anämie oder das begleitende Pepper-Syndrom: diffuse Leberschädigung mit schwerer Hepatomegalie und Atemnotsyndrom.
Das retroperitoneale Neuroblastom oder retroperitoneale Neuroblastom bei Kindern beginnt während seines Wachstums auf die Blase oder den Darm zu drücken, was zu Problemen beim Wasserlassen oder Stuhlgang sowie zu Schwellungen der Beine (bei Jungen schwillt der Hodensack an) führen kann.
Ein Neuroblastom des Mediastinums bei Kindern (mediastinales Neuroblastom) drückt häufig auf die obere Hohlvene und kann Schwellungen im Gesicht, am Hals, an den Armen und im oberen Brustbereich verursachen (mit bläulich-roter Hautverfärbung und subkutanen Knötchen). Husten und Keuchen, Atembeschwerden (Kurzatmigkeit) oder Schluckbeschwerden (Dysphagie) treten auf; vergrößerte Lymphknoten sind im Hals, oberhalb des Schlüsselbeins und in den Achselhöhlen zu beobachten.
Durch die Ausbreitung von Tumorzellen ins Knochenmark kommt es zu Anämie, Thrombozytopenie und Leukopenie mit Blutungsneigung.
Und bei Metastasen im periorbitalen Bereich treten dunkle Ringe oder Blutergüsse um die Augen auf. Ein solcher Tumor kann auch Kopfschmerzen und Schwindel, Exophthalmie (Vorwölbung der Augäpfel) und aufgrund der Kompression der Nervenenden herabhängende Augenlider (Ptosis) und eine Verkleinerung der Pupillen (Miosis) verursachen.
Ein abdominales Neuroblastom oder Neuroblastom der Bauchhöhle bei Kindern führt zur Bildung tastbarer Verschlüsse im Bauchraum, dessen Ausdehnung, Appetitlosigkeit, Verstopfung und erhöhtem Blutdruck. Ein Tumor, der auf das Rückenmark oder die Nervenwurzel drückt, kann zu Taubheit und Schwäche der Gliedmaßen sowie zur Unfähigkeit zu stehen, zu krabbeln oder zu gehen führen. Sind Knochen betroffen, können Knochenschmerzen auftreten.
Bei einem Tumor im Stadium 3-4 in der Bauchhöhle mit Lymphknotenschädigung können Tumorzellen in das Nierenparenchym eindringen, woraufhin sich bei Kindern ein ausgedehntes Neuroblastom der Niere entwickelt, das zu Funktionsstörungen der Niere führt.
Bühnen
- Das Neuroblastom im Stadium 1 ist ein Primärtumor, der auf einen Bereich des Körpers beschränkt ist; die Lymphknoten auf beiden Seiten sind nicht betroffen.
- Neuroblastom im Stadium 2. Im Stadium 2A ist der Primärtumor lokal begrenzt, aber groß; beidseitige Lymphknoten sind nicht betroffen. Im Stadium 2B sind die Lymphknoten auf der tumorbefallenen Körperseite metastasenpositiv.
- Neuroblastom Stadium 3: Der Primärtumor durchquert das Rückenmark oder die Mittellinie des Körpers, ein- oder beidseitige Metastasen finden sich in den Lymphknoten.
- Neuroblastom im Stadium 4: Der Tumor hat sich auf entfernte Lymphknoten, Knochenmark, Knochen, Leber oder andere Organe ausgebreitet. Stadium 4S wird bei Kindern unter einem Jahr mit einem lokalisierten Primärtumor mit Ausbreitung auf Haut, Leber oder Knochenmark festgestellt.
Internationales Neuroblastom-Risiko-Staging-System (INRGSS)
INRGSS verwendet bildgebend definierte Risikofaktoren (IDRFs). Dabei handelt es sich um Faktoren, die bei bildgebenden Untersuchungen erkennbar sind und dazu führen können, dass sich ein Tumor schwerer entfernen lässt.
INRGSS unterteilt Neuroblastome in 4 Stadien:
- L1: Der Tumor hat sich nicht von seinem Ursprungsort aus ausgebreitet und ist nicht in lebenswichtige Strukturen eingedrungen. Er ist auf einen Körperteil wie Hals, Brust oder Bauch beschränkt.
- L2: Der Tumor hat sich nicht weit von seinem Ausgangspunkt ausgebreitet (metastasiert) (beispielsweise kann er von der linken Seite des Bauchs auf die linke Seite der Brust gewachsen sein), aber er weist mindestens eine IDRF auf.
- M: Der Tumor hat in einen entfernten Körperteil metastasiert (ausgenommen Tumoren im MS-Stadium).
- MS: Metastatische Erkrankung bei Kindern unter 18 Monaten, bei der sich der Krebs nur auf die Haut, die Leber und/oder das Knochenmark ausgebreitet hat.
Komplikationen und Konsequenzen
Das Neuroblastom ist durch Komplikationen und Folgen gekennzeichnet wie:
- Ausbreitung (Metastasierung) in Lymphknoten, Knochenmark, Leber, Haut und Knochen;
- Kompression des Rückenmarks (die Schmerzen verursachen und zu Lähmungen führen kann);
- Entwicklung eines paraneoplastischen Syndroms (aufgrund der Wirkung bestimmter vom Tumor abgesonderter Chemikalien sowie des von seinen Zellen exprimierten Antigens Disialogangliosid GD2), das sich durch schnelle unwillkürliche Augenbewegungen, Koordinationsstörungen, Muskelkrämpfe und Durchfall äußert;
- Rückfälle nach Abschluss der Primärtherapie (wie die klinische Praxis zeigt, kommt es bei Hochrisiko-Neuroblastomen in 50 % der Fälle zu einem Rückfall).
Diagnose Neuroblastome
Zur Diagnose eines vermuteten Neuroblastoms bei einem Kind sind Untersuchungen, Labortests und bildgebende Verfahren erforderlich.
Blut- und Urinuntersuchungen werden auf Katecholamin (Noradrenalin und Dopamin) sowie Homovanillinsäure bzw. Vanillylmandelsäure (gebildet beim Stoffwechsel dieser Hormone) durchgeführt; außerdem wird ein Bluttest auf neurospezifische Enolase durchgeführt, ein Enzymimmunoassay (ELISA) des Blutserums durchgeführt und eine Knochenmarkanalyse durchgeführt (eine Probe wird durch Aspirationspunktion entnommen). Ein DNA-Test wird durchgeführt, um Mutationen festzustellen, und eine Biopsie wird zur zytomorphologischen Untersuchung des Tumorgewebes durchgeführt.
Nach der Entnahme der Biopsieproben werden diese in ein Labor geschickt, wo sie von einem Pathologen (einem Arzt mit Spezialausbildung in der Erkennung von Krebszellen) unter dem Mikroskop untersucht werden. Häufig werden auch spezielle Laboruntersuchungen durchgeführt, um festzustellen, ob es sich bei dem Tumor um ein Neuroblastom handelt.
Handelt es sich um ein Neuroblastom, können Laboruntersuchungen auch dabei helfen, festzustellen, wie schnell der Tumor wächst oder sich ausbreitet und welche Behandlung am besten wirkt.
Die instrumentelle Diagnostik visualisiert das Neoplasma mittels Ultraschall, Röntgen, MRT oder CT, PET mit Einführung von 18F-Fluordesoxyglucose oder MIBG-Scanning – Szintigraphie mit Metaiodbenzylguanidin. [ 1 ]
Differenzialdiagnose
Zu den Differentialdiagnosen gehören benignes Ganglioneurom, Ganglioneuroblastom, Rhabdomyosarkom und Nephroblastom.
Wen kann ich kontaktieren?
Behandlung Neuroblastome
Bei einem Neuroblastom hängt die Behandlung von der Risikogruppe des Patienten (Stadium des Tumorprozesses), der Lokalisation des Tumors, den genomischen Merkmalen der Tumorzellen und dem Alter des Kindes ab. Sie kann Überwachung, Operation, Chemotherapie, Strahlentherapie, Immuntherapie und hämatopoetische Stammzelltransplantation umfassen.
Die neoadjuvante oder adjuvante (prä- oder postoperative) Chemotherapie bei Neuroblastomen im Kindesalter erfolgt wie jede Chemotherapie bei Krebs in Zyklen: Das Medikament wird mehrere Tage hintereinander verabreicht, gefolgt von einer Pause zur Erholung des Körpers. Die Zyklen werden in der Regel alle drei bis vier Wochen wiederholt.
Folgende Medikamente (und deren Kombinationen) werden verwendet: Cyclophosphamid, Cisplatin oder Carboplatin, Doxorubicin (Adriamycin), Vincristin, Etoposid.
Zu den häufigsten Nebenwirkungen von Chemotherapeutika zählen Haarausfall, Appetitlosigkeit, Müdigkeit, Übelkeit und Erbrechen, Mundgeschwüre, Durchfall und Verstopfung. Chemotherapie kann das Knochenmark schädigen und zu einer Verringerung der Blutzellzahl führen.
Bei der zielgerichteten Immuntherapie (gerichtet gegen das Tumorantigen GD2) kommen Medikamente aus der Gruppe der monoklonalen Antikörper (Anti-GD2 MAb) Dinutuximab (Unituxin) und Naxitamab zum Einsatz. Sie werden intravenös mittels Langzeitinfusion in Kombination mit Granulozyten-Makrophagen-Kolonie-stimulierendem Faktor (Zytokin GM-CSF) und Interleukin-2 verabreicht.
Zu den Nebenwirkungen dieser Medikamente zählen Schmerzen (oft sehr starke), niedriger Blutdruck, erhöhter Puls, Kurzatmigkeit (mit möglicher Schwellung der Atemwege), erhöhte Temperatur, Übelkeit, Erbrechen und Durchfall sowie Veränderungen der Zell- und Mineralzusammensetzung des Blutes.
Um das Risiko eines erneuten Krebsausbruchs nach einer hochdosierten Chemotherapie und Stammzelltransplantation zu verringern, werden Kinder mit einem Hochrisiko-Neuroblastom mit systemischen Retinoiden, 13-cis-Retinsäure (Isotretinoin), behandelt. [ 2 ]
Chirurgische Behandlung des Neuroblastoms – Tumorentfernung, zum Beispiel offene Adrenalektomie oder laparoskopische Resektion des Nebennierenneuroblastoms; Lymphektomie (Entfernung der betroffenen Lymphknoten) usw. [ 3 ]
Bei einem Hochrisiko-Neuroblastom kann eine Strahlentherapie eingesetzt werden.[ 4 ]
Verhütung
Angesichts der Ursachen des Neuroblastoms bei Kindern kann die einzige vorbeugende Maßnahme eine genetische Beratung bei der Planung einer Schwangerschaft sein. Es sollte jedoch berücksichtigt werden, dass dieser Tumor nur in 1-2% der Fälle mit vererbten Mutationen assoziiert ist.
Prognose
Das infantile Neuroblastom hat die Fähigkeit, sich spontan zurückzubilden.
Prognosemarker
- Hochrisikotumoren sowie Neuroblastome bei Kindern aller Altersgruppen und aller Stadien (außer Stadium 4S) – mit erhöhter Expression des N-MYC-Gens und Amplifikation des N-Myc-Onkogens – haben eine ungünstige Prognose, die die Lebenserwartung beeinträchtigt.
- Tumorzellen, denen bestimmte Teile der Chromosomen 1 oder 11 fehlen (bekannt als 1p- oder 11q-Deletionen), haben eine schlechtere Prognose. Auch ein zusätzlicher Teil des Chromosoms 17 (17q-Gewinn) ist mit einer schlechteren Prognose verbunden.
- Neuroblastomzellen mit einer großen Menge an DNA haben eine bessere Prognose, insbesondere bei Kindern unter 2 Jahren.
- Neuroblastome, die über mehr Neurotrophin-Rezeptoren verfügen, insbesondere über den Nervenwachstumsfaktor-Rezeptor TrkA, haben eine bessere Prognose.
Überleben nach Risikogruppe der Childhood Oncology Group (COG)
- Niedrigrisikogruppe: Kinder der Niedrigrisikogruppe haben eine 5-Jahres-Überlebensrate von über 95 %.
- Mittlere Risikogruppe: Kinder der mittleren Risikogruppe haben eine 5-Jahres-Überlebensrate von 90 % bis 95 %.
- Hochrisikogruppe: Kinder der Hochrisikogruppe haben eine 5-Jahres-Überlebensrate von etwa 50 %.
Etwa 15 % der Krebstodesfälle im Kindesalter sind auf ein Neuroblastom zurückzuführen. Die Langzeitüberlebenschancen bei dieser Hochrisiko-Malignität liegen bei höchstens 40 %. Die Gesamtüberlebensrate nach fünf Jahren beträgt 67–74 %, in der Altersgruppe der Ein- bis Vierjährigen 43 % und bei einem im ersten Lebensjahr diagnostizierten Neuroblastom über 80 %.
Использованная литература