Facharzt des Artikels

Neue Veröffentlichungen
Achondroplasie
Zuletzt überprüft: 12.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
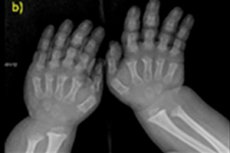
Es gibt viele seltene angeborene Krankheiten, und eine davon ist eine Verletzung des Knochenwachstums – Achondroplasie, die zu einer schweren unverhältnismäßigen Kleinwüchsigkeit führt.
Im Abschnitt über Entwicklungsanomalien des ICD-10 lautet der Code für diese Art der hereditären osteochondralen Dysplasie mit Wachstumsstörungen der Röhrenknochen und der Wirbelsäule Q77.4 [ 1 ]
Epidemiologie
In Bezug auf die Prävalenz der Achondroplasie sind statistische Daten aus verschiedenen Studien nicht eindeutig. Einige behaupten, dass diese Anomalie bei einem von 10.000 Neugeborenen auftritt, andere - bei einem von 26.000 bis 28.000 und wieder andere - bei 4-15 von 100.000 Fällen. [ 2 ]
Es gibt auch Informationen, dass bei über 50-jährigen Vätern die Häufigkeit einer Achondroplasie bei Kindern bei einem Fall pro 1875 Neugeborenen liegt.
Ursachen Achondroplasie
Die Ursache der Achondroplasie ist eine Verletzung der Osteogenese, insbesondere eine der Arten der intrauterinen Ossifikation der Diaphysen der Röhrenknochen des Skeletts – die enchondrale Ossifikation, bei der Knorpel in Knochengewebe umgewandelt wird. Weitere Informationen finden Sie unter - Knochenentwicklung und -wachstum
Eine Störung der Verknöcherung langer Knochen, d. h. eine fetale Achondroplasie, entsteht durch Mutationen im Gen der Membrantyrosinkinase – Fibroblasten-Wachstumsfaktor-Rezeptor 3 (FGFR3 auf Chromosom 4p16.3), die das Zellwachstum und die Zelldifferenzierung beeinflussen. Das Vorhandensein von FGFR3-Mutationen ist mit genetischer Instabilität und Veränderungen der Chromosomenzahl (Aneuploidie) verbunden.
Achondroplasie wird autosomal-dominant an ein Kind vererbt, d. h. es erhält eine Kopie des mutierten Gens (das dominant ist) und ein normales Gen auf einem Paar nicht-geschlechtlicher (autosomaler) Chromosomen. Daher ist die Vererbung dieses Defekts autosomal-dominant, und die Anomalie kann sich bei 50 % der Nachkommen manifestieren, wenn eine Kombination von Allelen dieses Gens (Genotyp) gekreuzt wird.
Darüber hinaus können Mutationen sporadisch auftreten, und wie die Praxis zeigt, werden Kinder mit Achondroplasie in 80 % der Fälle von Eltern mit normaler Körpergröße geboren.
Risikofaktoren
Die Hauptrisikofaktoren für die Geburt von Kindern mit Achondroplasie sind erblich bedingt. Leidet ein Elternteil an dieser Anomalie, liegt die Wahrscheinlichkeit, ein krankes Kind zu bekommen, bei 50 %; leiden beide Elternteile an dieser Anomalie, liegt sie ebenfalls bei 50 %, allerdings besteht ein 25-prozentiges Risiko für eine homozygote Achondroplasie, die zum Tod vor der Geburt oder im frühen Säuglingsalter führt.
Mit zunehmendem Alter des Vaters (näher an 40 Jahren und älter) steigt das Risiko einer Neumutation (De-novo-Mutation) des FGFR3-Gens.
Pathogenese
Bei der Erklärung der Pathogenese der Achondroplasie betonen Experten die Bedeutung des Transmembranproteins Tyrosin-Proteinkinase (kodiert durch das Gen FGFR3) bei der Regulierung der Teilung, Differenzierung und Apoptose von Zellen des Knorpelgewebes der Wachstumsfugen – Chondrozyten – sowie der normalen Entwicklung des Skeletts – Osteogenese und Mineralisierung des Knochengewebes.
Während der Embryonalentwicklung werden die Rezeptoren des Fibroblasten-Wachstumsfaktors 3 bei Vorhandensein einer Genmutation aktiver. Die Zunahme ihrer Funktionen stört die Übertragung zellulärer Signale und die Interaktion des extrazellulären Teils dieses Proteins mit Polypeptid-Fibroblasten-Wachstumsfaktoren (FGF). Infolgedessen tritt ein Fehler auf: Das Proliferationsstadium der Knorpelzellen wird kürzer und ihre Differenzierung beginnt früher als erwartet. All dies führt zu einer fehlerhaften Bildung und Fusion der Schädelknochen und zu Skelettdysplasie – einer Abnahme der langen Knochen, die mit ausgeprägter Kleinwüchsigkeit oder Zwergwuchs einhergeht.
Und zwei Drittel der Fälle von Kleinwuchs stehen im Zusammenhang mit Achondroplasie.
Symptome Achondroplasie
Abnormes Knochenwachstum verursacht klinische Symptome einer Achondroplasie wie:
- ausgeprägter Kleinwuchs (disproportionaler Zwergwuchs) mit einer durchschnittlichen Körpergröße im Erwachsenenalter von 123–134 cm;
- Verkürzung der proximalen Teile der unteren und oberen Extremitäten bei relativ normaler Rumpfgröße;
- verkürzte Finger und Zehen;
- vergrößerter Kopf (Makro- oder Megalozephalie); [ 3 ]
- spezifische Gesichtsmerkmale in Form einer vorstehenden Stirn und einer Hypoplasie des mittleren Gesichtsteils – ein eingesunkener Nasenrücken.
- schmaler kraniozervikaler Übergang. Einige Säuglinge mit Achondroplasie sterben im ersten Lebensjahr an Komplikationen im Zusammenhang mit dem kraniozervikalen Übergang. Bevölkerungsstudien deuten darauf hin, dass dieses erhöhte Sterberisiko ohne Untersuchung und Intervention bis zu 7,5 % betragen kann.[ 4 ]
- Funktionsstörungen des Mittelohrs sind häufig problematisch [ 5 ] und können bei unzureichender Behandlung zu Schallleitungsschwerhörigkeit führen, die so schwerwiegend ist, dass sie die Sprachentwicklung beeinträchtigt. Mehr als die Hälfte der Kinder benötigt einen Druckausgleichsschlauch. [ 6 ] Insgesamt leiden etwa 40 % der Menschen mit Achondroplasie an einem funktionell signifikanten Hörverlust. Auch die expressive Sprachentwicklung ist oft verzögert, wobei die Stärke des Zusammenhangs zwischen Hörverlust und expressiven Sprachproblemen fraglich ist.
- Eine Verbiegung des Schienbeins kommt bei Menschen mit Achondroplasie sehr häufig vor. Über 90 % der unbehandelten Erwachsenen weisen eine Verbiegung auf. [ 7 ] Bei der „Verbiegung“ handelt es sich um eine komplexe Deformität, die aus einer Kombination von seitlicher Neigung, Innentorsion des Schienbeins und dynamischer Instabilität des Knies resultiert. [ 8 ]
Säuglinge mit Achondroplasie zeichnen sich durch eine Muskelhypotonie aus, aufgrund derer sie später Bewegungsfähigkeiten erlernen und laufen. Intelligenz und kognitive Fähigkeiten sind von dieser Entwicklungsstörung nicht betroffen. [ 9 ], [ 10 ]
Folgen und Komplikationen
Diese Art der hereditären osteochondralen Dysplasie ist durch folgende Komplikationen und Folgen gekennzeichnet:
- wiederkehrende Ohrenentzündungen;
- obstruktive Schlafapnoe;
- Hydrozephalus;
- Fehlbiss und schiefe Zähne:
- Deformation der Beine (Varus oder Valgus) mit Gangänderung;
- hypertrophierte Lordose der Lendenwirbelsäule oder deren Krümmung (thorakolumbale Kyphose oder lumbale Skoliose) – mit Rückenschmerzen beim Gehen;
- Gelenkschmerzen (aufgrund einer falschen Knochenposition oder einer Kompression der Nervenwurzeln);
- Spinalkanalstenose und Rückenmarkkompression; Die häufigste medizinische Beschwerde im Erwachsenenalter ist eine symptomatische Spinalkanalstenose der L1-L4-Region. Die Symptome reichen von einer intermittierenden, reversiblen Claudicatio intermittens nach körperlicher Belastung bis hin zu schweren, irreversiblen Beinfunktionsstörungen und Harnverhalt.[ 11 ] Claudicatio intermittens und Stenose können sowohl sensorische (Taubheitsgefühl, Schmerzen, Schweregefühl) als auch motorische (Schwäche, Stolpern, eingeschränkte Gehfähigkeit) Symptome verursachen. Die vaskuläre Claudicatio intermittens entsteht durch Schwellungen der Blutgefäße nach Stehen und Gehen und ist durch Ruhe vollständig reversibel. Bei der Spinalkanalstenose handelt es sich um eine Läsion des Rückenmarks oder der Nervenwurzel durch den stenotischen Knochen des Wirbelkanals, und die Symptome sind irreversibel. Lokalisierte Symptome in einem bestimmten Dermatom können durch eine Stenose bestimmter Nervenwurzelforamina bedingt sein.
- Eine Verkleinerung der Brustwand mit eingeschränktem Lungenwachstum und verminderter Lungenfunktion (schwere Atemnot). Im Säuglingsalter leidet eine kleine Gruppe von Menschen mit Achondroplasie unter restriktiven Lungenproblemen. Kleine Brüste und eine erhöhte Brustcompliance führen zusammen zu einer reduzierten Lungenkapazität und einer restriktiven Lungenerkrankung [ 12 ].
Andere orthopädische Probleme
- Gelenkschwäche. Die meisten Gelenke sind im Kindesalter hypermobil. Im Allgemeinen hat dies kaum Auswirkungen, mit Ausnahme einer Knieinstabilität bei manchen Menschen.
- Diskoider Außenmeniskus: Diese kürzlich identifizierte strukturelle Anomalie kann bei manchen Menschen zu chronischen Knieschmerzen führen.[ 13 ]
- Arthritis: Eine konstitutive Aktivierung von FGFR-3, wie bei Achondroplasie, kann vor der Entwicklung von Arthritis schützen.[ 14 ]
- Acanthosis nigricans tritt bei etwa 10 % der Menschen mit Achondroplasie auf.[ 15 ] In dieser Population spiegelt es weder eine Hyperinsulinämie noch eine Malignität wider.
Die homozygote Achondroplasie, verursacht durch biallelische pathogene Varianten am Nukleotid 1138 des FGFR3-Gens, ist eine schwere Erkrankung mit qualitativ anderen radiologischen Befunden als bei der Achondroplasie. Frühe Todesfälle sind auf respiratorische Insuffizienz aufgrund einer zu kleinen Brustwand und neurologische Defizite aufgrund einer zervikomedullären Stenose zurückzuführen [Hall 1988].
Diagnose Achondroplasie
Bei den meisten Patienten wird die Diagnose einer Achondroplasie anhand charakteristischer klinischer Symptome und radiologischer Befunde gestellt. Bei Säuglingen oder bei fehlenden Symptomen werden genetische Tests, wie z. B. eine Karyotypanalyse, zur endgültigen Diagnose herangezogen.[ 16 ]
Im Rahmen der pränatalen Diagnostik können mittels molekulargenetischer Methoden Analysen des Fruchtwassers oder eine Chorionzottenprobe durchgeführt werden.
Anzeichen einer Achondroplasie im Ultraschall des Fötus – Verkürzung der Gliedmaßen und typische Gesichtszüge – werden nach der 22. Schwangerschaftswoche sichtbar.
Zur instrumentellen Diagnostik gehören auch Röntgenaufnahmen des Skeletts oder Ultraschalluntersuchungen der Knochen. Eine Röntgenaufnahme bestätigt die Diagnose anhand von Daten wie einem großen Schädel mit einem engen Hinterhauptloch und einer relativ kleinen Basis, kurzen Röhrenknochen und verkürzten Rippen, kurzen und abgeflachten Wirbelkörpern, einem verengten Wirbelkanal und einer Verkleinerung der Beckenschaufeln.
Differenzialdiagnose
Eine Differentialdiagnose mit Hypophysen-Zwergwuchs, angeborener spondyloepiphysärer und diastrophischer Dysplasie, Hypochondroplasie, Shereshevsky-Turner- und Noonan-Syndrom sowie Pseudoachondroplasie ist erforderlich. Der Unterschied zwischen Pseudoachondroplasie und Achondroplasie besteht darin, dass bei Patienten mit Zwergwuchs bei Pseudoachondroplasie die Kopfgröße und die Gesichtszüge normal sind.
Wen kann ich kontaktieren?
Behandlung Achondroplasie
Empfehlungen zur Behandlung von Kindern mit Achondroplasie wurden vom Ausschuss für Genetik der American Academy of Pediatrics herausgegeben. Diese Empfehlungen dienen als Orientierungshilfe und ersetzen nicht die individuelle Entscheidungsfindung. Eine aktuelle Übersichtsarbeit [Pauli & Botto 2020] enthält ebenfalls Leitlinien. Es gibt Fachkliniken, die auf die Behandlung von Skelettdysplasie spezialisiert sind; ihre Empfehlungen können geringfügig von diesen allgemeinen Empfehlungen abweichen.
Zu den Empfehlungen gehören (unter anderem) die folgenden.
Hydrozephalus. Wenn Anzeichen oder Symptome eines erhöhten Hirndrucks auftreten (z. B. beschleunigtes Kopfwachstum, dauerhaft vorgewölbte Fontanelle, auffällige Vergrößerung oberflächlicher Venen im Gesicht, Reizbarkeit, Erbrechen, Sehstörungen, Kopfschmerzen), ist die Überweisung an einen Neurochirurgen erforderlich.
Die vermutete Ätiologie des Hydrozephalus bei Achondroplasie ist ein erhöhter intrakranieller Venendruck aufgrund einer Stenose der Foramina jugularis. Daher ist die Standardbehandlung ein ventrikuloperitonealer Shunt. Eine endoskopische dritte Ventrikulostomie kann jedoch bei manchen Patienten hilfreich sein,[ 17 ] was darauf hindeutet, dass andere Mechanismen, wie z. B. eine Obstruktion des vierten Ventrikelausgangs aufgrund einer kraniozervikalen Stenose, beteiligt sein können.[ 18 ]
Stenose des kraniozervikalen Übergangs. Beste Prädiktoren für die Notwendigkeit einer subokzipitalen Dekompression:
- Hyperreflexie oder Klonus der unteren Extremitäten
- Zentrale Hypopnoe bei Polysomnographie
- Verringerung der Größe des Foramen magnum, ermittelt durch Computertomographie des kraniozervikalen Übergangs und verglichen mit den Normen für Kinder mit Achondroplasie.[ 19 ]
- Als weiterer Faktor, der bei der Entscheidung für eine Operation berücksichtigt werden sollte, wurden kürzlich Anzeichen einer Rückenmarkkompression und/oder T2-gewichtete Signalanomalien vorgeschlagen.
Bei deutlichen Anzeichen einer symptomatischen Kompression sollte dringend eine Überweisung an einen pädiatrischen Neurochirurgen zur Dekompressionsoperation erfolgen. [ 20 ]
Die Behandlung von obstruktiver Schlafapnoe kann Folgendes umfassen:
- Adenotonsillektomie
- Positiver Atemwegsdruck
- Tracheostomie in Extremfällen
- Gewichtsverlust
Diese Eingriffe können zu einer Verbesserung der Schlafstörungen und einer gewissen Verbesserung der neurologischen Funktionen führen.[ 21 ]
In seltenen Fällen, in denen die Obstruktion so schwerwiegend ist, dass eine Tracheotomie erforderlich ist, wurde eine Mittelgesichtsvorverlagerung durchgeführt, um die Obstruktion der oberen Atemwege zu beheben.[ 22 ]
Funktionsstörung des Mittelohrs. Häufige Mittelohrentzündungen, anhaltende Flüssigkeitsansammlungen im Mittelohr und daraus resultierender Hörverlust sollten bei Bedarf aggressiv behandelt werden. Langzeitröhrchen werden empfohlen, da sie oft bis zum siebten oder achten Lebensjahr benötigt werden.[ 23 ]
Bei auftretenden Problemen empfiehlt sich in jedem Alter der Einsatz entsprechender Behandlungsmethoden.
Kleinwuchs. Mehrere Studien haben die Wachstumshormontherapie (GH) als mögliche Behandlung für Achondroplasie aufgrund von Kleinwuchs untersucht.[ 24 ]
Insgesamt zeigen diese und andere Reihen eine anfängliche Beschleunigung des Wachstums, der Effekt lässt jedoch mit der Zeit nach.
Im Durchschnitt ist mit einer Körpergrößenzunahme von nur etwa 3 cm im Erwachsenenalter zu rechnen.
Eine erweiterte Beinverlängerung mittels verschiedener Techniken ist für manche Patienten weiterhin möglich. Ein Größenzuwachs von bis zu 30–35 cm ist möglich. [ 25 ] Komplikationen sind häufig und können schwerwiegend sein.
Während einige dafür plädieren, diese Verfahren bereits im Alter von sechs bis acht Jahren durchzuführen, plädieren viele Kinderärzte, klinische Genetiker und Ethiker dafür, solche Operationen aufzuschieben, bis ein junger Mensch in der Lage ist, eine fundierte Entscheidung zu treffen.
Zumindest in Nordamerika entscheidet sich nur ein kleiner Teil der Betroffenen für eine fortgeschrittene Gliedmaßenverlängerung. Der medizinische Beirat der Little People of America hat eine Stellungnahme zur Anwendung fortgeschrittener Gliedmaßenverlängerungen herausgegeben.
Adipositas: Maßnahmen zur Vorbeugung von Adipositas sollten bereits im frühen Kindesalter beginnen. Standardbehandlungen gegen Adipositas sollten bei Menschen mit Achondroplasie wirksam sein, obwohl der Kalorienbedarf geringer ist. [ 26 ]
Zur Verlaufsverfolgung sollten speziell für Achondroplasie entwickelte Standard-Gewichts- und -Größendiagramme verwendet werden. Wichtig zu beachten ist, dass diese Kurven keine perfekten Gewicht-Größen-Kurven darstellen; sie wurden aus Tausenden von Datenpunkten von Menschen mit Achondroplasie abgeleitet.
Es wurden Standards für den Body-Mass-Index (BMI) für Kinder im Alter von 16 Jahren und jünger entwickelt. [ 27 ] Für Erwachsene mit Achondroplasie ist der BMI nicht standardisiert; Vergleiche mit BMI-Kurven für die durchschnittliche Körpergröße führen zu irreführenden Ergebnissen. [ 28 ]
Varusdeformität. Jährliche orthopädische Nachuntersuchungen durch einen mit Achondroplasie vertrauten Arzt oder einen orthopädischen Chirurgen werden empfohlen. Kriterien für chirurgische Eingriffe wurden veröffentlicht.[ 29 ]
Bei progressiver symptomatischer Verkrümmung ist eine Überweisung an einen Orthopäden erforderlich. Eine asymptomatische Varusdeformität selbst erfordert in der Regel keine chirurgische Korrektur. Verschiedene Interventionen stehen zur Auswahl (z. B. gesteuertes Wachstum mit acht Platten, Valgusosteotomie und Derotationsosteotomie). Es gibt keine kontrollierten Studien, die die Ergebnisse der Behandlungsoptionen vergleichen.
Kyphose. Säuglinge mit Achondroplasie entwickeln häufig eine flexible Kyphose. Es gibt ein Protokoll, um die Entwicklung einer fixierten Winkelkyphose zu verhindern. Dazu gehört der Verzicht auf flexible Kinderwagen, Schaukeln und Babytragen. Es wird vom ungestützten Sitzen abgeraten; beim Halten des Babys immer Gegendruck auf den Rücken ausüben.
- Bei den meisten Kindern bessert sich die Kyphose deutlich oder verschwindet ganz, wenn sie eine orthograde Haltung einnehmen und mit dem Gehen beginnen. [ 30 ]
- Bei Kindern, bei denen nach Steigerung der Rumpfstärke und Beginn des Gehens keine spontane Remission eintritt, ist eine Orthese in der Regel ausreichend, um ein Fortbestehen der thorakolumbalen Kyphose zu verhindern.[ 31 ]
- Wenn eine schwere Kyphose anhält, kann eine Wirbelsäulenoperation erforderlich sein, um neurologische Komplikationen zu verhindern.[ 32 ]
Spinalkanalstenose: Wenn schwere Anzeichen und/oder Symptome einer Spinalkanalstenose auftreten, ist eine dringende Überweisung an einen chirurgischen Spezialisten erforderlich.
In der Regel wird eine erweiterte und weite Laminektomie empfohlen. Die Relevanz des Eingriffs hängt von der Höhe (z. B. thorakal oder lumbal) und dem Grad der Stenose ab. Patienten zeigten bessere Ergebnisse und eine verbesserte Funktion, je früher sie nach Symptombeginn operiert wurden [ 33 ].
Impfungen: Die Achondroplasie schließt alle Routineimpfungen nicht aus. Angesichts des erhöhten Atemwegsrisikos sind DTaP-, Pneumokokken- und Grippeimpfungen besonders wichtig.
Anpassungsbedarf: Aufgrund der geringen Körpergröße können Anpassungen der Umgebung notwendig sein. In der Schule können dazu Hocker, tiefer angebrachte Lichtschalter, Toiletten in geeigneter Höhe oder andere barrierefreie Einrichtungen, niedrigere Tische und Fußstützen vor den Stühlen gehören. Alle Kinder sollten im Notfall das Gebäude selbstständig verlassen können. Kleine Hände und schwache Sehnen können die Feinmotorik beeinträchtigen. Geeignete Anpassungen sind beispielsweise eine kleinere Tastatur, beschwerte Stifte und glattere Schreibflächen. Die meisten Kinder sollten einen IEP- oder 504-Plan haben.
Zum Fahren sind fast immer Pedalverlängerungen erforderlich. Auch Arbeitsplatzanpassungen wie niedrigere Tische, kleinere Tastaturen, Stufen und Toilettenzugänge können erforderlich sein.
Sozialisierung: Aufgrund der deutlich spürbaren Kleinwüchsigkeit, die mit der Achondroplasie einhergeht, können betroffene Personen und ihre Familien Schwierigkeiten haben, soziale Kontakte zu knüpfen und sich an die Schule zu gewöhnen.
Selbsthilfegruppen wie Little People of America, Inc (LPA) können Familien dabei helfen, diese Probleme durch gegenseitige Unterstützung, persönliche Beispiele und Programme zur sozialen Sensibilisierung anzugehen.
Informationen zu Beschäftigung, Bildung, Behindertenrechten, Adoption kleinwüchsiger Kinder, medizinischen Fragen, angemessener Kleidung, adaptiven Geräten und Kindererziehung sind über einen nationalen Newsletter sowie Seminare und Workshops verfügbar.
Es gibt keine Medikamente oder nicht-medikamentöse Behandlung, die diesen angeborenen Defekt heilen kann.
Am häufigsten wird physikalische Therapie eingesetzt; eine Behandlung kann auch bei Hydrozephalus (mittels Shunt oder endoskopischer Ventrikulostomie), Fettleibigkeit, [ 34 ] Apnoe, [ 35 ] Mittelohrentzündung oder Spinalkanalstenose erforderlich sein.
In einigen Kliniken werden nach Erreichen des fünften bis siebten Lebensjahres des Kindes chirurgische Eingriffe vorgenommen: Verlängerung der Knochen der Schienbeine, Oberschenkel und sogar der Schulterknochen oder Korrektur der Deformität – mit Hilfe von Operationen und speziellen orthopädischen Geräten – in drei bis vier Schritten, die jeweils bis zu 6–12 Monate dauern.
Therapie unter Untersuchung
Die Gabe eines C-Typ-natriuretischen Peptidanalogon befindet sich derzeit in klinischen Studien. Erste Ergebnisse zeigen eine gute Verträglichkeit und eine Steigerung der Wachstumsgeschwindigkeit gegenüber dem Ausgangswert bei Kindern mit Achondroplasie ( Studienort ). [ 36 ] Konjugiertes C-Typ-natriuretisches Peptid befindet sich derzeit ebenfalls in klinischen Studien. [ 37 ] Weitere Überlegungen betreffen die Tyrosinkinasehemmung [ 38 ], Meclizin [ 39 ] und einen löslichen rekombinanten humanen FGFR3-Decoy. [ 40 ]
Durchsuchen Sie clinicaltrials.gov in den USA und das EU Clinical Trials Registry in Europa nach Informationen zu klinischen Studien für ein breites Spektrum an Krankheiten und Beschwerden.
Verhütung
Die einzige vorbeugende Maßnahme ist die pränatale Diagnostik angeborener Erkrankungen. [ 41 ], [ 42 ]
Prognose
Wie lange leben Menschen mit Achondroplasie? Etwa 10 Jahre weniger als die durchschnittliche Lebenserwartung.
Da pathologische Veränderungen des Knochengewebes und der Gelenke zu Einschränkungen der Selbstversorgung und Mobilität führen, wird Kindern mit dieser Diagnose der Status einer Behinderung zuerkannt. Langfristig haben die meisten Patienten eine normale Prognose, mit zunehmendem Alter steigt jedoch das Risiko für Herzerkrankungen. [ 43 ]