Facharzt des Artikels

Neue Veröffentlichungen
Hämophagozytisches Syndrom bei Kindern: primär, sekundär
Zuletzt überprüft: 23.04.2024

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
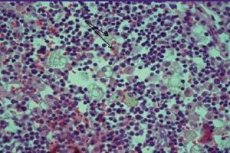
Eine seltene und komplexe Erkrankung - das Hämophagozytose-Syndrom - wird als hämophagozytische Lymphogystyozytose bezeichnet. Diese schwere Erkrankung ist mit der Entstehung eines Mangels an vielen Organen infolge einer unkontrollierten Aktivierung der Effektorkomponente der zellulären Immunabwehr verbunden.
Beim hämophagozytischen Syndrom kommt es zu einer starken Zunahme der Funktionalität toxischer T-Lymphozyten und Makrophagen, was zur Entwicklung zahlreicher entzündungshemmender Zytokine führt. Dieser Prozess wiederum beinhaltet eine intensive systemische Entzündungsreaktion und eine Störung der Funktion vieler Organe im großen Maßstab.
Ursachen hämophagozytisches Syndrom
Das Hämophagozytäre Syndrom ist öfter primär - dh, erblich bedingt infolge der genetischen Verwirrung bei der Arbeit der Makrophage.
Das sekundäre hämophagozytische Syndrom wird auch als erworben bezeichnet: es ist mit verschiedenen Infektionskrankheiten, Tumorprozessen, Autoimmunerkrankungen, angeborenen Stoffwechselstörungen assoziiert.
In der klassischen Version des hereditären Typs des Hämophagozytose-Syndroms werden Kinder häufig zur Behandlung auf der Intensivstation oder Intensivstation in infektiösen Krankenhäusern zur Diagnose von septischen Komplikationen oder intrauterinen generalisierten Infektionen eingesetzt. Die direkte Diagnose des hämophagozytischen Syndroms wird oft nach einem tödlichen Ausgang festgestellt.
Auf den ersten Blick jedoch können Infektionskrankheiten viralen oder mikrobiellen Ursprungs eine solche Komplikation wie ein lebensbedrohliches hämophagozytisches Syndrom verursachen.
Das hämophagozytische Syndrom bei Erwachsenen ist in den meisten Fällen fast immer sekundär: Am häufigsten entwickelt sich die Pathologie vor dem Hintergrund lymphoproliferativer Erkrankungen und chronischer VEB-Infektionen.
Hämophagozytische Syndrom bei Kindern kann sowohl primäre als auch sekundäre - aufgrund der übertragenen Infektionskrankheiten (Windpocken, Meningoenzephalitis, etc.) sein.
Symptome hämophagozytisches Syndrom
Anzeichen für das Syndrom wurden erstmals Mitte des letzten Jahrhunderts beschrieben. Folgende charakteristische Symptome wurden identifiziert:
- stabiles Fieber;
- eine Abnahme der Menge an hämatopoetischen Substanzen;
- eine Vergrößerung der Leber und der Milz;
- ausdrucksvolles hämorrhagisches Syndrom.
Die Patienten können das Phänomen der Leberversagen, hohe Konzentrationen von Ferritin und transaminazina, ein klares Bild einer neurologischen Störung des ZNS funktionellen, ein hohes Maß an Triglyceriden im Serum beobachten, beschleunigt die Blutgerinnung und Gerinnungsstörung.
Häufig finden die Erkrankten vergrößerte Lymphknoten, Hautausschläge, Gelbfärbung der Sklera, Haut und Schleimhäute sowie Schwellungen.
Milzparenchym, sinusoidale Leberkapillaren, Sinus der Lymphknoten, Knochenmark und Zentralnervensystem zeichnen sich durch diffuse Infiltration aktiver Makrophagen vor dem Hintergrund der hämophagozytischen Symptome aus. Lymphdrüsengewebe ist erschöpft. Bei der Untersuchung der Leber gibt es Läsionen, die für die chronische Form der persistierenden Entzündung typisch sind.
Formen
Es gibt zwei klinische Formen, die zunächst schwer zu unterscheiden sind.
- Primäre hämophagozytische Lymphohistiozytose, bei der es sich um eine autosomal-rezessive Pathologie handelt, bei deren Entwicklung die Mutation des Perforin-Gens von primärer Bedeutung ist.
- Sekundäre Form der hämophagozytischen Lymphohistiozytose, die als Folge einer übermäßigen Immunaktivität der Kette der mononukleären Phagozyten entsteht.
Komplikationen und Konsequenzen
- Beitritt der Infektion mit nachfolgender Intoxikation. Diese Komplikation ist gekennzeichnet durch einen allmählichen Funktionsverlust der Hauptorgane und -systeme, Fieber, Erschöpfung des Patienten.
- Bösartige Degeneration von Zellen. Malignität ist in der Regel die Entwicklung von Lymphomen, Leukämien und anderen bösartigen Erkrankungen.
- Autoimmunpathologien - eine Art aggressive Reaktion der eigenen Immunabwehr des Patienten.
- Anhaltender Rückgang der Immunität mit der Entwicklung von Immundefizienz-Zustand.
- Mangel an Nieren- und Leberfunktion.
- Innere Blutung, Blutung.
- Der Tod des Patienten vor totaler Organdysfunktion oder vor septischen Komplikationen.
Diagnose hämophagozytisches Syndrom
Wenn die Familiengeschichte nicht belastet ist, dann bestimmen Sie das primäre oder sekundäre Hämophagozytose-Syndrom ist sehr schwierig. Um eine genaue Diagnose zu stellen, ist es notwendig, eine histologische Differenzierung der Hämophagozytose durchzuführen.
Viele Krankheiten sind schwer zu bestimmen, indem nur die Informationen aus Gewebebiopsien verwendet werden: Lymphknoten, Leber und Knochenmark.
Die Durchführung von immunologischen Studien, die uns erlauben, die inhibierte Funktion von NK-Zellstrukturen zu sehen und den Gehalt des Interleukin-2-Rezeptors zu erhöhen, kann nicht als Grundlage für die Diagnose dienen. Zusätzlich werden die Merkmale des klinischen Bildes, Schädigung und Störung des Zentralnervensystems und Veränderungen der Blutzusammensetzung des Patienten berücksichtigt.
Der letzte Punkt in der Diagnose sind die Daten der molekulargenetischen Analyse.
Differenzialdiagnose
Die Differenzierung der Krankheit ist extrem schwierig, wobei der Ansatz in Abhängigkeit vom Alter des Patienten bestimmt werden muss. In der Pädiatrie ist es wichtig, die genetischen Formen des hämophagozytischen Syndroms so früh wie möglich zu erkennen und alle möglichen Faktoren zu analysieren, die auf eine erbliche Pathologie hinweisen können.
So ist die schnelle Entwicklung des Syndroms während der ersten 12 Lebensmonate mit einer unkomplizierten Familienanamnese typisch für die primäre Form des hämophagozytischen Syndroms. Die beobachtete Expression von Perforin in den NK-Zellstrukturen durch das Verfahren der Durchflusszytofluorometrie und molekulargenetische Studien von Perforin helfen, die richtigen Diagnose in ungefähr 30% der Fälle von erblichem hemophagocytic Syndrom zu etablieren. Mit solchen Syndromen wird das gleichzeitige Erscheinen der Erkrankung auf dem Hintergrund des Albinismus aufgedeckt:
Wenn die Vererbung einen X-chromosomalen Typ hat, das heißt, wenn sich die Krankheit in verwandten Männchen entlang der Linie der Mutter entwickelt, dann ist das Vorhandensein eines autoimmunen lymphoproliferativen Syndroms am wahrscheinlichsten.
Bei sekundärem hämophagozytischem Syndrom gilt es vor allem maligne Tumore frühzeitig zu erkennen, die im Erwachsenenalter häufig die Ursache des Syndroms sind.
Wen kann ich kontaktieren?
Behandlung hämophagozytisches Syndrom
Die Behandlung des hämophagozytischen Syndroms ist ziemlich kompliziert: Der Erfolg einer solchen Behandlung hängt weitgehend vom Alter des Patienten und davon ab, wie rechtzeitig die Krankheit entdeckt wurde.
Therapeutische Regime für Hämophagozytose-Syndrom beinhalten die Verwendung von Glukokortikosteroiden (Dexamethason), Zytostatika (Etoposid, Ciclosporin A). Zytotoxische Arzneimittel werden verschrieben, um die entzündungsfördernde Wirkung von Phagozyten durch weitere Alogen-Transplantation von Stammzellen zu unterdrücken.
Ein einzelnes Behandlungsschema für das hämophagozytische Syndrom wurde noch nicht bestimmt. Eine etiotrope Behandlung wird als nicht ausreichend zur Bekämpfung des Syndroms angesehen, und die Verwendung von Immunsuppressoren kann den Verlauf des virus-bakteriellen Prozesses nachteilig beeinflussen.
Es wird empfohlen, hohe Dosen von Immunglobulin zu injizieren, bezogen auf die Menge von 1-2 mg pro Kilogramm des Patientengewichts pro Tag.
Plasmaphoresis kann als Teil einer pathogenetischen Behandlung zur Überwachung von Hypercytokinämie verschrieben werden.
Die Basis der Behandlung ist Splenektomie und Transplantation des Spenderknochenmarks.
Verhütung
Experten haben derzeit keine klaren Informationen über Methoden zur Prävention des primären hämophagozytischen Syndroms, da die Ursachen für diese Pathologie nicht vollständig geklärt sind.
Was das sekundäre hämophagozytische Syndrom anbelangt, können vorbeugende Maßnahmen wie folgt aussehen:
- kompetente und rechtzeitige Behandlung von viralen und mikrobiellen Infektionen;
- qualifizierte Behandlung von Autoimmunpathologien unter Aufsicht eines Facharztes für rheumatologische Untersuchungen.
Prognose
Die Prognose des hämophagozytischen Syndroms wird als äußerst ungünstig angesehen, was aus den statistischen Daten ersichtlich ist: sechs Todesfälle unter sieben Fällen. Die maximale Überlebensdauer beträgt derzeit zwei Jahre.
Das Hämophagozyten-Syndrom gilt als eine sehr komplexe und heimtückische Krankheit, die heute mit Ausnahme der Infektion mit dem Immunschwächevirus "konkurriert" und die Häufigkeit der Folgen sogar noch übertrifft.
 [27]
[27]