Facharzt des Artikels

Neue Veröffentlichungen
T-Zell-Lymphome der Haut
Last reviewed: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
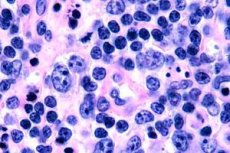
Am häufigsten werden T-Zell-Lymphome bei älteren Menschen registriert, obwohl vereinzelte Fälle der Erkrankung auch bei Kindern beobachtet werden. Männer erkranken doppelt so häufig wie Frauen. T-Zell-Lymphome sind epidermotroper Natur.
Ursachen T-Zell-Lymphome der Haut
Ursachen und Pathogenese kutaner T-Zell-Lymphome sind noch nicht vollständig geklärt. Derzeit betrachten die meisten Forscher das humane T-Zell-Leukämievirus Typ 1 (HTLV-1) I als den wichtigsten ätiologischen Faktor für die Entwicklung maligner T-Zell-Lymphome der Haut. Darüber hinaus wird die Rolle anderer Viren bei der Entstehung von T-Zell-Lymphomen diskutiert: Epstein-Barr-Virus, Herpes simplex Typ 6. Bei Patienten mit T-Zell-Lymphom finden sich Viren in der Haut, im peripheren Blut und in den Langerhans-Zellen. Antikörper gegen HTLV-I werden bei vielen Patienten mit Mycosis fungoides nachgewiesen.
Eine wichtige Rolle in der Pathogenese von T-Zell-Lymphomen spielen immunpathologische Prozesse in der Haut, von denen der wichtigste die unkontrollierte Proliferation klonaler Lymphozyten ist.
Von Lymphozyten, Epithelzellen und Zellen des Makrophagensystems produzierte Zytokine haben entzündungsfördernde und proliferative Wirkungen (IL-1, verantwortlich für die Lymphozytendifferenzierung; IL-2 – T-Zell-Wachstumsfaktor; IL-4 und IL-5, die den Zustrom von Eosinophilen in die Läsion und deren Aktivierung usw. erhöhen). Durch den Zustrom von T-Lymphozyten in die Läsion bilden sich Pautrier-Mikroabszesse. Gleichzeitig mit der Zunahme der Lymphozytenproliferation wird die Aktivität von Antitumor-Abwehrzellen unterdrückt: natürliche Killerzellen, lymphozytotoxische Lymphozyten, dendritische Zellen, insbesondere Langerhans-Zellen, sowie Zytokine (IL-7, IL-15 usw.) – Tumorwachstumshemmer. Die Rolle erblicher Faktoren kann nicht ausgeschlossen werden. Das Auftreten familiärer Fälle und der häufige Nachweis einiger Histokompatibilitätsantigene (HLA B-5 und HLA B-35 – bei hochmalignen Hautlymphomen, HLA A-10 – bei weniger aggressiven Lymphomen, HLA B-8 – bei der erythrodermischen Form der Mycosis fungoides) bestätigen die erbliche Natur der Dermatose.
Klinische Beobachtungen deuten auf eine mögliche Transformation chronischer Dermatosen (Neurodermitis, atopische Dermatitis, Psoriasis usw.) in eine Mycosis fungoides hin. Der Schlüsselfaktor ist die langfristige Persistenz von Lymphozyten im Entzündungsherd, die die Immunüberwachung stören und die Entstehung eines Klons maligner Lymphozyten und damit die Entwicklung eines malignen proliferativen Prozesses fördern.
Die Einwirkung physikalischer Faktoren auf den Körper, wie Sonneneinstrahlung, ionisierende Strahlung und chemische Substanzen, kann zur Entstehung eines Klons „genotraumatischer“ Lymphozyten führen, die eine mutagene Wirkung auf Lymphzellen haben und zur Entwicklung maligner Lymphozyten führen.
Daher können T-Zell-Lymphome als multifaktorielle Erkrankung betrachtet werden, die mit der Aktivierung von Lymphozyten unter dem Einfluss verschiedener karzinogener, „genotraumatisierender“ Faktoren und der Entstehung eines dominanten T-Zell-Klons beginnt. Der Schweregrad der Immunüberwachungsstörung und der Klon maligner Lymphozyten bestimmen die klinischen Manifestationen (Fleck-, Plaque- oder Tumorelemente) von T-Zell-Lymphomen.
Pathogenese
Im Frühstadium der Mycosis fungoides werden Akanthose mit breiten Fortsätzen, Hyperplasie und Verdichtung basaler Keratinozyten, vakuoläre Degeneration einiger Basalzellen, atypische Mitosen in verschiedenen Schichten der Epidermis, Epidermotropismus des Infiltrats mit Eindringen von Lymphozyten in die Epidermis beobachtet. In der Dermis werden um die Gefäße herum kleine Infiltrate beobachtet, die aus einzelnen mononukleären Zellen mit hyperchromen Kernen bestehen – „mykotischen“ Zellen. Im zweiten Stadium kommt es zu einer Zunahme des dermalen Infiltrats und eines Epidermotropismus der Infiltratzellen, wodurch maligne Lymphozyten in die Epidermis eindringen und Cluster in Form von Potrier-Mikroabszessen bilden. Im dritten, tumorösen Stadium, kommt es zu massiver Akanthose und leichter Atrophie der Epidermis sowie zu einer verstärkten Infiltration der Epidermis durch Tumorlymphozyten, die multiple Potrier-Mikroabszesse bilden. Das massive Infiltrat erstreckt sich über die gesamte Dicke der Dermis und bedeckt einen Teil der Hypodermis. Es werden Blastenformen von Lymphozyten beobachtet.
Kutanes großzelliges anaplastisches T-Zell-Lymphom
Es handelt sich um eine Gruppe lymphoproliferativer Prozesse, die durch das Vorhandensein von Proliferaten atypischer klonaler großer anaplastischer CD30+ T-Zellen gekennzeichnet sind. In der Regel entwickelt es sich sekundär im Tumorstadium der Mycosis fungoides oder beim Sézary-Syndrom, kann sich aber auch unabhängig oder mit der Verbreitung systemischer Lymphome dieses Typs entwickeln. Klinisch entsprechen solche Lymphome der sogenannten dekapitierten Form der Mycosis fungoides in Form einzelner oder mehrerer Knoten, meist gruppiert.
Histologisch nimmt das Proliferat nahezu die gesamte Dermis ein, mit oder ohne Epidermotropismus im Falle einer epidermalen Atrophie.
Zytologisch können Tumorzellen in Größe und Form variieren. Basierend auf diesen Eigenschaften unterscheidet man zwischen mittel- und großzelligen pleomorphen T-Zell-Lymphomen mit Kernen unterschiedlicher unregelmäßiger Konfiguration – gewunden, mehrlappig, mit dichtem Chromatin, einem gut definierten Nukleolus und relativ reichlich Zytoplasma; immunoblastisch – mit großen runden oder ovalen Kernen mit klarem Karyoplasma und einem zentral gelegenen Nukleolus; anaplastisch – mit hässlichen, sehr großen Zellen mit Kernen unregelmäßiger Konfiguration und reichlich Zytoplasma. Phänotypisch gehört diese gesamte Gruppe zu den T-Helfer-Lymphomen und kann CD30+ oder CD30- sein.
R. Willemze et al. (1994) zeigten, dass der Verlauf des CD30+ Lymphoms günstiger ist. Genotypisch lässt sich eine klonale Umlagerung des T-Lymphozytenrezeptors nachweisen.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ], [ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ]
Symptome T-Zell-Lymphome der Haut
Die häufigste Erkrankung in der Gruppe der T-Zell-Lymphome der Haut ist die Mycosis fungoides, die etwa 70 % der Fälle ausmacht. Es gibt drei klinische Formen der Erkrankung: klassisch, erythrodermisch und enthauptet. T-Zell-Lymphome sind durch Polymorphismus von Hautausschlägen in Form von Flecken, Plaques und Tumoren gekennzeichnet.
Die erythrodermische Form der Mycosis fungoides beginnt in der Regel mit unkontrollierbarem Juckreiz, Schwellungen, allgemeiner Hyperämie und dem Auftreten von erythematös-squamösen Läsionen an der Haut des Rumpfes und der Extremitäten, die innerhalb von 1–2 Monaten verschmelzen und eine Erythrodermie entwickeln. Fast alle Patienten weisen eine palmar-plantare Hyperkeratose und eine diffuse Ausdünnung des Haares auf der gesamten Haut auf. Alle Lymphknotengruppen sind stark vergrößert. Vergrößerte inguinale, femorale, axilläre und kubale Lymphknoten werden als „Pakete“ von dichter, elastischer Konsistenz ertastet, nicht mit dem umgebenden Gewebe verwachsen und schmerzlos. Der Allgemeinzustand verschlechtert sich stark: Fieber mit einer Körpertemperatur von bis zu 38–39 °C, Nachtschweiß, Schwäche und Gewichtsverlust treten auf. Derzeit wird das Sézary-Syndrom von vielen Dermatologen als die seltenste leukämische Variante der erythrodermischen Form der Mycosis fungoides angesehen.
Im Lymphozytogramm wird eine ausgeprägte Leukozytose festgestellt – Sézary-Zellen. Sézary-Zellen sind maligne T-Helferzellen, deren Kerne eine gefaltete zerebriforme Oberfläche mit tiefen Einstülpungen der Kernmembran aufweisen. Ein tödlicher Ausgang wird nach 2–5 Jahren festgestellt, häufige Ursachen sind Herz-Kreislauf-Erkrankungen und Intoxikationen.
Die dekapitierte Form der Mycosis fungoides ist durch die schnelle Entwicklung tumorartiger Läsionen auf scheinbar gesunder Haut ohne vorherige langfristige Plaquebildung gekennzeichnet. Diese Form ist durch einen hohen Malignitätsgrad gekennzeichnet, der als Manifestation eines Lymphosarkoms gilt. Innerhalb eines Jahres wird ein tödlicher Ausgang beobachtet.
Bühnen
Die klassische Form der Mycosis fungoides ist durch drei Entwicklungsstadien gekennzeichnet: Erythematös-Plattenepithel, Plaque und Tumor.
Das erste Stadium ähnelt dem klinischen Bild einiger gutartiger entzündlicher Dermatosen – Ekzem, seborrhoischer Dermatitis, Plaque-Parapsoriasis. In diesem Stadium der Erkrankung werden Flecken unterschiedlicher Größe beobachtet, intensiv rosa, rosarot mit violetter Tönung, runde oder ovale Umrisse mit relativ klaren Grenzen, oberflächliche kleieartige oder feinplättchenförmige Abschuppung. Die Elemente befinden sich oft an verschiedenen Hautstellen, am häufigsten am Rumpf und im Gesicht. Allmählich nimmt ihre Zahl zu. Mit der Zeit kann der Prozess den Charakter einer Erythrodermie annehmen (erythrodermisches Stadium). Der Ausschlag kann jahrelang bestehen bleiben oder spontan verschwinden. Anders als bei gutartigen entzündlichen Dermatosen sind die Elemente des Ausschlags und des Juckreizes in diesem Stadium therapieresistent.
Das infiltrativ-plaqueartige Stadium entwickelt sich über mehrere Jahre. Anstelle der zuvor vorhandenen fleckigen Hautausschläge treten runde oder unregelmäßig geformte, intensiv violette Plaques auf, die sich deutlich von der gesunden Haut abgrenzen, dicht sind und eine schuppige Oberfläche aufweisen. Ihre Konsistenz ähnelt „dicker Pappe“. Einige von ihnen bilden sich spontan zurück und hinterlassen dunkelbraune Hyperpigmentierungen und/oder Atrophie (Poikilodermie). Der Juckreiz ist in diesem Stadium noch intensiver und schmerzhafter, Fieber und Gewichtsverlust treten auf. In diesem Stadium kann eine Lymphadenopathie auftreten.
Im dritten, dem Tumorstadium, erscheinen schmerzlose Tumoren von dichter, elastischer Konsistenz und gelbroter Farbe, die sich aus Plaques entwickeln oder auf scheinbar gesunder Haut entstehen. Die Form der Tumoren ist kugelig oder abgeflacht und ähnelt oft einem Pilzhut. Tumoren können überall auftreten. Ihre Anzahl variiert stark von einzeln bis zu Dutzenden, die Größe von 1 bis 20 cm im Durchmesser. Wenn sich lange bestehende Tumoren auflösen, bilden sich Geschwüre mit unebenen Rändern und einem tiefen Boden, die die Faszie oder den Knochen erreichen. Am häufigsten sind Lymphknoten, Milz, Leber und Lunge betroffen. Der Allgemeinzustand verschlechtert sich, Vergiftungssymptome treten auf und verstärken sich, Schwäche entwickelt sich. Die durchschnittliche Lebenserwartung von Patienten mit der klassischen Form der Mycosis fungoides ab dem Zeitpunkt der Diagnose beträgt 5 bis 10 Jahre. Todesfälle werden normalerweise durch interkurrente Erkrankungen beobachtet: Lungenentzündung, Herz-Kreislaufversagen, Amyloidose. Subjektiv ist Juckreiz zu spüren, und wenn sich Tumoren auflösen, treten Schmerzen in den betroffenen Bereichen auf.
Was muss untersucht werden?
Behandlung T-Zell-Lymphome der Haut
Im erythematös-squamösen Stadium benötigen die Patienten keine Antitumortherapie; ihnen werden topische Kortikosteroide (Prednisolon, Betamethason, Dexamethason-Derivate), Interferon alpha (3 Millionen IE täglich, dann 3-mal wöchentlich für 3-6 Monate, abhängig von den klinischen Manifestationen oder der Wirksamkeit der Behandlung), Interferon gamma (100.000 IE täglich für 10 Tage, der Zyklus wird 12-3-mal mit einer 10-tägigen Pause wiederholt), PUVA-Therapie oder Re-PUVA-Therapie verschrieben. Die Wirksamkeit der PUVA-Therapie beruht auf der selektiven Bildung kovalenter Vernetzungen von Psoralenen mit DNA in proliferierenden T-Helferzellen, die deren Teilung hemmen. In der zweiten Phase werden zusätzlich zu den oben genannten Wirkstoffen systemische Kortikosteroide (30–40 mg Prednisolon täglich für 1,5–2 Monate) und Zytostatika (Prospedin 100 mg täglich, insgesamt 4–5 Injektionen) eingesetzt. Die Kombination von Interferonen mit anderen Therapiemethoden hat einen ausgeprägteren therapeutischen Effekt (Interferone + PUVA, Interferone + Zytostatika, Interferone + aromatische Retinoide).
Im Tumorstadium ist die Polychemotherapie die wichtigste Methode. Es wird eine Kombination aus Vincristin (0,5–1 mg intravenös einmal täglich, insgesamt 4–5 Injektionen) mit Prednisolon (40–60 mg täglich oral während der Chemotherapie), Prospidin (100 mg täglich, insgesamt 3 g) und Interferonen eingesetzt. Empfohlen werden photodynamische Therapie, Elektronenstrahltherapie und Photopherese (extrakorporale Photochemotherapie).