Facharzt des Artikels

Neue Veröffentlichungen
Keratoderma: Ursachen, Symptome, Diagnose, Behandlung
Zuletzt überprüft: 07.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
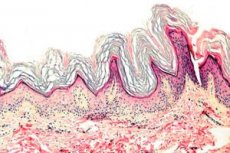
Keratodermie ist eine Gruppe von Dermatosen, die durch eine Störung des Verhornungsprozesses gekennzeichnet ist – übermäßige Hornbildung, hauptsächlich an den Handflächen und Fußsohlen.
Die Ursachen und die Pathogenese der Erkrankung sind noch nicht vollständig geklärt. Untersuchungen haben ergeben, dass Keratodermien durch Mutationen in den Genen verursacht werden, die für Keratin 6, 9 und 16 kodieren. Vitamin-A-Mangel, hormonelle Störungen, vor allem der Geschlechtsdrüsen, sowie bakterielle und virale Infektionen spielen eine wichtige Rolle in der Pathogenese. Sie sind ein Symptom von Erbkrankheiten und Tumoren der inneren Organe (parapsoriatische Keratodermien).
Symptome. Man unterscheidet zwischen diffuser (Unna-Tost-Keratodermie, Meleda-Keratodermie, Papillon-Lefevre-Keratodermie, mutilierende Keratodermie und Syndrome, bei denen die diffuse Keratodermie eines der Hauptsymptome ist) und fokaler (disseminierte gefleckte Keratodermie Fischer-Buschke, Akrokeratoelastoidose Kosti, begrenzte Keratodermie Bruhauer-Franzesthesti, lineare Keratodermie Fuchs usw.) Keratodermie.
Die Winy-Tost-Keratodermie (Synonyme: kongenitale Ichthyose der Handflächen und Fußsohlen, Winy-Tost-Syndrom) wird autosomal-dominant vererbt. Es kommt zu einer diffusen übermäßigen Verhornung der Haut der Handflächen und Fußsohlen (manchmal nur der Fußsohlen), die sich in den ersten beiden Lebensjahren entwickelt. Der pathologische Hautprozess beginnt mit einer leichten Verdickung der Haut der Handflächen und Fußsohlen in Form eines lividen Erythemstreifens an der Grenze zur gesunden Haut. Mit der Zeit erscheinen auf ihrer Oberfläche glatte, gelbliche Hornschichten. Die Läsion breitet sich selten auf die Handrücken oder Finger aus. Bei einigen Patienten können sich oberflächliche oder tiefe Risse bilden und es wird eine lokale Hyperhidrose festgestellt. Bei dem vom Autor beobachteten Patienten litten der Onkel mütterlicherseits, sein Bruder und sein Sohn an einer Winy-Tost-Keratodermie.
Es werden Fälle von Schäden an Nägeln (Verdickung), Zähnen und Haaren bei der Winy-Tost-Keratodermie in Kombination mit verschiedenen Skelettanomalien und Erkrankungen der inneren Organe sowie des Nerven- und Hormonsystems beschrieben.
Histopathologie. Histologisch zeigen sich ausgeprägte Hyperkeratose, Granulose, Akanthose und kleine entzündliche Infiltrate in der oberen Dermis. Differenzialdiagnose: Die Erkrankung muss von anderen Keratodermieformen abgegrenzt werden.
Die Meleda-Keratodermie (Synonyme: Meleda-Krankheit, kongenitales progressives Akrokeratom, Siemens-palmoplantare transgradiente Keratose, Kogoy-hereditäre palmoplantare progressive Keratose) wird autosomal-rezessiv vererbt. Diese Form der Keratodermie ist durch dicke, gelbbraune Hornschichten mit tiefen Rissen gekennzeichnet. An den Rändern der Läsion ist ein mehrere Millimeter breiter violett-violetter Rand sichtbar. Der Prozess breitet sich typischerweise auf Hand- und Fußrücken, Unterarme und Schienbeine aus. Die meisten Patienten leiden unter lokaler Hyperhidrose. Dabei werden die Oberflächen der Handflächen und Fußsohlen leicht feucht und mit schwarzen Punkten (Schweißdrüsengängen) bedeckt.
Die Krankheit kann sich im Alter zwischen 15 und 20 Jahren entwickeln. Die Nägel verdicken sich und verformen sich.
Histopathologie. Die histologische Untersuchung zeigt eine Hyperkeratose, manchmal Akanthose, und ein chronisch entzündliches Infiltrat in der papillären Dermis.
Differenzialdiagnose: Die Melela-Keratodermie muss von der Unna-Tost-Keratodermie unterschieden werden.
Keratoderma Papillon-Lefevre (Synonym: Palmoplantare Hyperkeratose mit Parodontitis) wird autosomal-rezessiv vererbt.
Die Krankheit manifestiert sich im 2.-3. Lebensjahr. Das klinische Bild der Krankheit ähnelt der Melela-Krankheit. Darüber hinaus sind Veränderungen an den Zähnen charakteristisch (Auffälligkeiten beim Durchbruch der Milch- und bleibenden Zähne mit der Entwicklung von Karies, Gingivitis, schnell fortschreitender Parodontose mit vorzeitigem Zahnverlust).
Histopathologie. Die histologische Untersuchung zeigt eine Verdickung aller Schichten der Epidermis, insbesondere der Hornschicht, sowie unbedeutende Zellhaufen von Lymphozyten und Histiozyten in der Dermis.
Differentialdiagnose. Die Erkrankung muss von anderen Keratosen unterschieden werden. Ein wichtiges Unterscheidungsmerkmal ist die charakteristische Zahnpathologie, die bei anderen Formen hereditärer diffuser Keratosen nicht auftritt.
Keratoderma mutilans (Synonyme: Fonwinkel-Syndrom, hereditäres mutilierendes Keratom) ist eine Form der diffusen Keratodermie, die autosomal-dominant vererbt wird. Sie entwickelt sich im zweiten Lebensjahr und ist durch diffuse Hornablagerungen an Handflächen und Fußsohlen mit Hyperhidrose gekennzeichnet. Mit der Zeit bilden sich strangartige Rillen an den Fingern, was zu Kontrakturen und spontaner Amputation der Finger führt. Follikuläre Keratose manifestiert sich auf dem Handrücken sowie im Bereich der Ellenbogen- und Kniegelenke. Die Nagelplatten sind verändert (oft uhrglasartig). Fälle von Hypogonadismus, Rubinalopezie, Hörverlust und Pachyonychie wurden beschrieben.
Histopathologie. Die histologische Untersuchung zeigt eine schwere Hyperkeratose, Granulose, Akanthose und kleine entzündliche Infiltrate in der Dermis, bestehend aus Lymphozyten und Histiozyten.
Bei der Abgrenzung der mutilierenden Keratodermie von anderen Formen der diffusen Keratodermie sollte vor allem der für andere Formen untypische Mutilationseffekt berücksichtigt werden. Bei der Differentialdiagnose aller Formen der diffusen Keratodermie muss berücksichtigt werden, dass sie eines der Hauptsymptome einer Reihe erblicher Syndrome sein kann.
Behandlung. Neotigazon ist in der allgemeinen Therapie der Keratodermie indiziert. Die Dosis des Arzneimittels richtet sich nach der Schwere des Prozesses und beträgt 0,3–1 mg/kg Körpergewicht des Patienten. Bei fehlendem Neotigazon wird Vitamin A in einer Dosis von 100 bis 300.000 mg pro Tag über einen längeren Zeitraum empfohlen. Die externe Therapie besteht in der Anwendung von Salben mit aromatischen Retinoiden, Keratolytika und Steroiden.
 [
[ Was bedrückt dich?
Was muss untersucht werden?
Wie zu prüfen?