Facharzt des Artikels

Neue Veröffentlichungen
T-Zell-Lymphom der Haut
Zuletzt überprüft: 23.04.2024

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
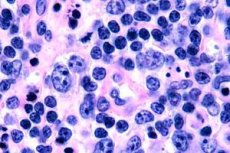
Meistens werden T-Zell-Lymphome bei älteren Menschen aufgezeichnet, obwohl es auch bei Kindern vereinzelte Fälle gibt. Männer sind doppelt so oft krank wie Frauen. T-Zell-Lymphome sind epidermotropisch.
Ursachen t-Zell-Lymphome der Haut
Die Ursachen und Pathogenese von T-Zell-Lymphomen sind nicht vollständig geklärt. Derzeit betrachten die meisten Forscher das menschliche T-Zell-Leukämie-Typ-1 (HTLV-1) I-Virus als den hauptsächlichen ätiologischen Faktor, der die Entwicklung von T-Zell-malignen Lymphomen der Haut initiiert. Daneben wird die Rolle anderer Viren bei der Entstehung von T-Zell-Lymphomen diskutiert: Epstein-Barr-Virus, Herpes simplex Typ 6. Bei Patienten mit T-Zell-Lymphom sind Viren in der Haut, peripheren Blut, Langerhans-Zellen gefunden. Antikörper gegen HTVL-I werden bei vielen Patienten mit Pilzmykose nachgewiesen.
Ein wichtiger Platz in der Pathogenese von T-Zell-Lymphomen spielen immunpathologische Prozesse in der Haut, deren Hauptursache die unkontrollierte Proliferation klonaler Lymphozyten ist.
Cytokine, die von Lymphozyten produziert, Epithelzellen und Zellen des Makrophagen-Systems hat pro-inflammatorische und proliferative Aktivität (IL-1, die für die Differenzierung von Lymphozyten, IL-2 - ein Faktor von T-Zellwachstum, IL-4 und IL-5, Verstärkungs Einströmen Läsionen Eosinophilen und ihre Aktivierung usw.). Als Ergebnis des Einströmens in die Läsion von T-Lymphozyten werden Mikroabszesse gebildet. Gleichzeitig mit der Erhöhung die Proliferation von Lymphozyten auftritt, Anti-Tumor-Aktivität Hemmung Zellen schützen: natürliche Killerzellen, lymphozytotoxischen Lymphozyten, dendritische Zellen, insbesondere von Langerhans-Zellen sowie Zytokine (IL-7, IL-15, etc.) - der Tumorwachstumshemmer. Die Rolle von Erbfaktoren ist nicht ausgeschlossen. Verfügbarkeit familiäre Fälle, häufige Entdeckung bestimmter Histokompatibilitäts-Antigene (HLA B-5 und HLA B-35 - ein hoher Grad der Bösartigkeit von Hautlymphom, HLA A-10 - Lymphome vorkommendes weniger aggressiv, HLA B-8 - wenn eritrodermicheskoy Form Mycosis fungoides) bestätigen die Erblichkeit der Dermatose.
Klinische Beobachtungen deuten auf eine mögliche Umwandlung von chronischen Langzeitdermatosen (Neurodermitis, atopische Dermatitis, Psoriasis etc.) in Pilzmykose hin. In diesem Fall ist der Schlüssel Langzeitpersistenz von Lymphozyten in Entzündung, die Immunüberwachung unterbrechen und zur Entstehung von malignen Lymphozyten Klon beitragen und damit die Entwicklung von bösartigem proliferativen Prozess.
Auswirkungen auf den Körper von physikalischen Faktoren wie Sonneneinstrahlung, ionisierende Strahlung, Chemikalien können clone „genotravmaticheskih“ Lymphozyten mutagene Wirkung auf Lymphoidzellen und die Entwicklung von Malignität von B-Lymphozyten verursachen.
Somit können T-Zell-Lymphome als multifaktorielle Erkrankung in Betracht gezogen werden, die mit der Aktivierung von Lymphozyten zu verschiedenen Karzinogenen ausgesetzt beginnen, „genotravmiruyuschih“ Faktoren und die Entstehung einer dominanten Zellklon T. Der Schweregrad der gestörten Immunüberwachung, ein Klon von malignen Lymphozyten, bestimmt die klinischen Manifestationen (Flecken, Plaque oder Tumorelemente) von T-Zell-Lymphomen.

Pathogenese
Im frühen Stadium des Mycosis fungoides markiert acanthosis mit breiten Prozessen, Verdichtung und Hyperplasie der basalen Keratinozyten vacuolar Degeneration des basalen Zellteils, atypischer Mitose in verschiedenen Schichten der Epidermis, epidermogropizm Infiltration mit Infiltration von Lymphozyten in den Epidermis. In den Dermis gibt es kleine infiltriert um die Gefäße, die aus isolierten mononuklearen Zellen mit -hyperchromasie, - „mykotische“ Zellen. In der zweiten Stufe gibt es die Schwere der dermalen Infiltration und Infiltration epidermotropizm Zellen erhöht, in einem malignen Lymphocyten dringen in die Epidermis resultierenden Anhäufungen in Form Mikroabszesse PONV bilden. In dem dritten, einen Tumor, eine massive gekennzeichnet Stufe Akanthose und ein leichte Atrophie der Epidermis, erhöht durch Lymphozyten epidermalen Tumorinfiltration, die mehrere Mikroabszesse PONV bilden. Massive Infiltration über die gesamte Dicke der Dermis und Hypodermis Erfassung Teil. Blasformen von Lymphozyten werden notiert.
Großzelliges anaplastisches T-Zell-Lymphom der Haut
Präsentiert von einer Gruppe von lymphoproliferativen Prozessen, die durch die Anwesenheit von Proliferaten aus atypischen klonalen großen anaplastischen CD30 + -T-Zellen charakterisiert sind. In der Regel entwickelt es sich sekundär im Tumorstadium der Pilzmykose oder mit dem Sie-Zary-Syndrom, jedoch kann es sich unabhängig oder bei der Verbreitung von systemischen Lymphomen dieses Typs entwickeln. Klinisch entsprechen solche Lymphome der sogenannten enthaupteten Form von Pilzmykose in Form von einzelnen oder mehreren Knoten, die gewöhnlich gruppiert sind.
Histologisch, Proliferate besetzen fast die gesamte Dermis mit oder ohne Epidermotropismus, wenn die Epidermis Atrophie ist.
Zytologisch können Tumorzellen in Größe und Form variieren. Auf der Grundlage dieser Eigenschaften gewonnen Mittel- und Groß pleomorph T-Zell-Lymphom mit Kernen variieren Fehlkonfiguration - konvolyutnymi, Multi-Blade, mit einem dichten Chromatin, eine ausgeprägten Kernkörperchen und eher reichlich Zytoplasma; immunoblastisch - mit großen runden oder ovalen Kernen mit einem erleuchteten Karyoplasma und einem zentral gelegenen Nukleolus; anaplastisch - mit hässlichen sehr großen Zellen mit unregelmäßig geformten Kernen und reichlich Zytoplasma. Phänotypisch gehört die gesamte Gruppe zu T-Helfer-Lymphomen und kann CD30 + oder CD30- sein.
R. Willemze et al. (1994) zeigten, dass der Verlauf des CD30 + -Lymphoms günstiger ist. Die klonale Reorganisation des T-Lymphozyten-Rezeptors wird genotypisch aufgedeckt.
[1], [2], [3], [4], [5], [6], [7], [8], [9], [10], [11], [12],
Symptome t-Zell-Lymphome der Haut
Die häufigste Erkrankung in der Gruppe der T-Zell-Lymphome der Haut ist Pilzmykose, die etwa 70% der Fälle ausmacht. Es gibt drei klinische Formen der Krankheit: klassisch, erythrodermisch und enthauptet. T-Zell-Lymphome sind durch Polymorphismus von Hautausschlägen in Form von Flecken, Plaques, Tumoren gekennzeichnet.
Erythrodermischer Form der Mycosis fungoides beginnt in der Regel mit unbändiger Juckreiz, Schwellung, Rötung universal, erscheint auf der Haut des Rumpfes und der Gliedmaßen gerötete-Plattenepithelkarzinome Läsionen , die erythroderma innerhalb von 1-2 Monaten fusionieren neigen und zu entwickeln. Praktisch alle Patienten haben eine palmar-plantare Hyperkeratose und eine diffuse Haarverdünnung in der gesamten Haut. Alle Gruppen von Lymphknoten sind stark vergrößert. Vergrößert inguinal, femoral, axillaren, cubital tastbare Lymphknoten in der Form von „Paketen“ plotnoelasticheskoy Konsistenz, nicht gelöteten umgebenden Gewebe, schmerzlos. Der Allgemeinzustand verschlechtert sich stark: Fieber mit Körpertemperatur bis 38-39 ° C, Nachtschweiß, Schwäche und Gewichtsverlust. Derzeit gilt das Cesary-Syndrom bei vielen Dermatologen als die seltenste Leukämievariante der erythrodermalen Form der Pilzmykose.
Es gibt eine ausgeprägte Leukozytose in Lymphozytogrammen - Cesari-Zellen. Cesari-Zellen sind bösartige T-Helfer, deren Kerne eine gefaltete Hirnoberfläche mit tiefen Invaginationen der Kernmembran haben. Der letale Ausgang wird nach 2-5 Jahren beobachtet, deren häufige Ursache kardiovaskuläre Pathologie und Intoxikation ist.
Die kopflose Form der Pilzmykose ist durch die rasche Entwicklung von tumorähnlichen Foci auf scheinbar gesunder Haut ohne vorherige langanhaltende Plaques gekennzeichnet. Diese Form zeichnet sich durch eine hohe Malignität aus, die als Manifestation des Lymphosarkoms angesehen wird. Das tödliche Ergebnis wird während des ganzen Jahres bemerkt.
Bühnen
Die klassische Form der Pilzmykose ist durch drei Entwicklungsstadien gekennzeichnet: erythematös-squamös, Plaque und Tumor.
Das erste Stadium ähnelt dem klinischen Bild einiger gutartiger entzündlicher Dermatosen - Ekzeme, seborrhoische Dermatitis, Plaque-Parapsoriasis. In diesem Krankheitsstadium gibt es Flecken unterschiedlicher Größe, intensiv rosa, rosa-rot mit einem violetten Farbton, runde oder ovale Umrisse, mit relativ klaren Grenzen, oberflächliche Schuppenbildung oder kleine Schuppenbildung. Elemente befinden sich oft an verschiedenen Stellen der Haut, häufiger an Rumpf und Gesicht. Allmählich erhöht sich die Anzahl von ihnen. Im Laufe der Zeit kann der Prozess die Art der Erythrodermie (erythrodermisches Stadium) annehmen. Hautausschläge können jahrelang bestehen oder spontan verschwinden. Im Gegensatz zu gutartigen entzündlichen Dermatosen sind Hautausschlag und Juckreiz in diesem Stadium gegen eine fortlaufende Therapie resistent.
Das infiltrative-bpley-Stadium entwickelt sich innerhalb einiger Jahre. An Stelle von vorbestehenden Makulaeruptionen erscheinen Plaques mit abgerundeten oder unregelmäßigen Konturen, intensiv violett, klar abgegrenzt von gesunder Haut, dicht, mit einer schälenden Oberfläche. Ihre Konsistenz ähnelt "dicker Karton". Einige von ihnen sind spontan aufgelöst und hinterlassen Bereiche von dunkelbrauner Hyperpigmentierung und / oder Atrophie (Poikilodermie). Juckreiz in diesem Stadium ist noch intensiver und schmerzhaft, Fieber wird festgestellt, Gewichtsverlust wird festgestellt. In diesem Stadium kann Lymphadenopathie beobachtet werden.
Im dritten Tumorstadium entstehen schmerzlose Tumore von dicht-elastischer Konsistenz mit gelb-roter Farbe, die sich aus Plaques entwickeln oder auf scheinbar gesunder Haut erscheinen. Die Form der Tumoren ist kugelförmig oder abgeflacht und ähnelt oft einer Pilzkappe. Tumore können überall auftreten. Die Anzahl von ihnen variiert stark von einzelnen bis zu Dutzenden, mit Größen von 1 bis 20 cm im Durchmesser. Bei der Auflösung von Langzeittumoren bilden sich Geschwüre mit unebenen Kanten und einem tiefen Boden, die bis zur Faszie oder den Knochen reichen. Die am häufigsten betroffenen Lymphknoten, Milz, Leber und Lunge. Der allgemeine Zustand verschlechtert sich, erscheint und wächst in das Phänomen der Vergiftung ein, entwickelt Schwäche. Die durchschnittliche Lebenserwartung von Patienten mit einer klassischen Form von Pilzmykosen ab dem Zeitpunkt der Diagnose beträgt 5 bis 10 Jahre. Die Sterblichkeit wird in der Regel bei chronischen Erkrankungen festgestellt: Lungenentzündung, Herz-Kreislauf-Insuffizienz, Amyloidose. Juckreiz ist subjektiv empfunden, und mit dem Zerfall von Tumoren - der Schmerz in den Läsionen.
Was muss untersucht werden?
Behandlung t-Zell-Lymphome der Haut
In Schritt gerötete-Plattenepithelkarzinomen Patienten, die die Anti-Tumor-Therapie nicht brauchten, bezeichnen sie als externe Kortikosteroide (Prednisolon-Derivate, Betamethason, Dexamethason), alpha-Interferon (3 Millionen täglich ME, gefolgt von 3-mal pro Woche für 3-6 Monate., Je nach den klinischen Manifestationen (oder Wirksamkeit der Behandlung), Interferon-gamma bei 100 000 ME pro Tag für 10 d., 12-3 mal der Zyklus wird wiederholt mit einem Intervall von 10 Tagen.), PUVA-Therapie oder Fe PUVA-Therapie. Die Wirksamkeit des Verfahrens der PUVA-Therapie ist auf die selektive Bildung von kovalenten Vernetzungen basierend auf Psoralen DNA in proliferierenden T-Helferzellen, die ihre Teilung hemmt. Im zweiten Schritt mit Ausnahme der obigen Mittel werden verwendet, systemische Kortikosteroide (30-40 mg Prednison pro Tag für 1,5-2 Monate), Zytostatika (prospedin 100 mg pro Tag täglich, 4-5 Injektionen in allen). Kombination mit anderen Methoden der Interferon-Therapie einen deutlicheren therapeutischen Effekt (+ PUVA Interferone, Interferone + Zytostatika, Interferone + aromatische Retinoide) aufweist.
Im Tumorstadium ist die Polychemotherapie die Hauptmethode. Tragen Sie eine Kombination von Vincristin (0,5-1 mg / einmal täglich, 4-5 Injektionen gesamt) mit Prednison (60 bis 40 mg täglich durch den Mund für einen Zeitraum von Chemotherapie) prospidina (100 mg pro Tag, insgesamt 3 g) Interferone. Empfohlene photodynamische, Elektronenstrahl-Therapie, Phototherapie (extrakorporale Photochemotherapie).