Facharzt des Artikels

Neue Veröffentlichungen
Usher-Syndrom
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Das Usher-Syndrom ist eine Erbkrankheit, die sich durch völlige Taubheit von Geburt an und fortschreitende Erblindung mit zunehmendem Alter äußert. Der Sehverlust ist mit einer Retinitis pigmentosa verbunden, einer Pigmentdegeneration der Netzhaut. Viele Menschen mit Usher-Syndrom leiden zudem unter schweren Gleichgewichtsstörungen.
Epidemiologie
Dank der Forschung konnte festgestellt werden, dass das Usher-Syndrom etwa 8 % der untersuchten taubstummen Kinder betrifft (die Tests wurden in speziellen Einrichtungen für Taubstumme durchgeführt). Eine Pigmentretinitis wurde bei 6–10 % der Patienten mit angeborener Taubheit beobachtet, die wiederum bei etwa 30 % der Menschen mit einer Pigmentretinitis auftritt.
Man geht davon aus, dass diese Krankheit bei etwa 3–10 von 100.000 Menschen weltweit auftritt. Sie kann bei Frauen und Männern gleichermaßen auftreten. Etwa 5–6 % der Weltbevölkerung leiden an diesem Syndrom. Etwa 10 % aller Fälle schwerer Schwerhörigkeit im Kindesalter sind auf das Usher-Syndrom Typ I und II zurückzuführen.
In den USA sind die Typen 1 und 2 am häufigsten. Zusammen machen sie etwa 90 bis 95 Prozent aller Fälle des Usher-Syndroms bei Kindern aus.
Ursachen Usher-Syndrom
Die Usher-Syndrome Typ I, II und III haben eine autosomal-rezessive Ursache, während Typ IV als X-Chromosomenstörung gilt. Die Ursachen für Blindheit und Taubheit, die bei diesem Syndrom auftreten, sind noch nicht ausreichend erforscht. Es wird angenommen, dass Betroffene überempfindlich auf Bestandteile reagieren, die die DNA-Struktur schädigen können. Darüber hinaus kann die Erkrankung mit Störungen des Immunsystems einhergehen, wobei in diesem Fall noch kein genaues Bild dieses Prozesses vorliegt.
1989 wurden bei Patienten mit Typ-II-Syndrom erstmals Chromosomenanomalien festgestellt. Dies könnte in Zukunft dazu führen, die Gene zu isolieren, die das Syndrom verursachen. Möglicherweise könnten diese Gene auch bei Trägern identifiziert und spezielle pränatale Gentests entwickelt werden.
 [ 8 ]
[ 8 ]
Risikofaktoren
Das Syndrom wird vererbt, wenn beide Elternteile betroffen sind, d. h. es wird rezessiv vererbt. Ein Kind kann die Krankheit auch erben, wenn seine Eltern Träger des Gens sind. Wenn beide zukünftigen Eltern dieses Gen haben, beträgt die Wahrscheinlichkeit, ein Kind mit diesem Syndrom zu bekommen, 1 zu 4. Eine Person, die nur ein Gen für das Syndrom hat, gilt als Träger, weist aber keine Symptome der Erkrankung auf. Heutzutage ist es noch nicht möglich festzustellen, ob eine Person das Gen für diese Krankheit hat.
Wenn ein Kind von Eltern geboren wird, von denen einer kein solches Gen besitzt, ist die Wahrscheinlichkeit, dass es das Syndrom erbt, sehr gering, aber es wird auf jeden Fall ein Träger sein.
Symptome Usher-Syndrom
Zu den Symptomen des Usher-Syndroms zählen Hörverlust und eine abnorme Ansammlung pigmentierter Zellen in den Augenstrukturen. Der Patient entwickelt daraufhin eine Degeneration der Netzhaut, die zu einer Verschlechterung des Sehvermögens und in den schwersten Fällen schließlich zum Sehverlust führt.
Ein sensorineuraler Hörverlust kann leicht oder vollständig sein und verschlimmert sich in der Regel von Geburt an nicht. Eine Netzhautpigmentkrankheit kann sich jedoch bereits im Kindesalter oder später entwickeln. Testergebnisse haben gezeigt, dass die zentrale Sehschärfe über viele Jahre erhalten bleiben kann, selbst wenn sich das periphere Sehen verschlechtert (ein sogenannter „Tunnelblick“).
Dies sind die Hauptsymptome der Krankheit, zu denen manchmal noch weitere Störungen hinzukommen können, wie etwa Psychosen und andere psychische Störungen, Probleme mit dem Innenohr und/oder Katarakte.
Formen
Im Laufe der Forschung wurden drei Typen dieser Krankheit sowie eine vierte Form identifiziert, die recht selten ist.
Typ I der Erkrankung ist durch angeborene Taubheit sowie Gleichgewichtsstörungen gekennzeichnet. Oft beginnen solche Kinder erst im Alter von 1,5 Jahren zu laufen. Die Sehverschlechterung beginnt in der Regel im Alter von 10 Jahren, und die endgültige Entwicklung der Nachtblindheit beginnt im Alter von 20 Jahren. Bei Kindern mit dieser Erkrankung kann es zu einer fortschreitenden Verschlechterung des peripheren Sehens kommen.
Bei Typ II liegt eine mittelschwere oder angeborene Taubheit vor. In diesem Fall tritt häufig keine Verschlechterung der partiellen Taubheit mehr auf. Die Pigmentretinitis entwickelt sich gegen Ende der Adoleszenz oder nach 20 Jahren. Nachtblindheit entwickelt sich in der Regel im Alter von 29 bis 31 Jahren. Die Sehbehinderung bei Typ II schreitet im Allgemeinen etwas langsamer voran als bei Typ I.
Typ III der Erkrankung ist durch einen fortschreitenden Hörverlust gekennzeichnet, der normalerweise während der Pubertät beginnt. Im gleichen Zeitraum (etwas später als der Hörverlust) entwickelt sich außerdem allmählich eine Retinitis pigmentosa, die zur Entwicklung einer fortschreitenden Erblindung beitragen kann.
Manifestationen der Pathologie Typ IV treten hauptsächlich bei Männern auf. In diesem Fall werden auch fortschreitende Störungen sowie Hör- und Sehverlust beobachtet. Diese Form ist sehr selten und hat in der Regel einen X-chromosomalen Charakter.
Diagnose Usher-Syndrom
Die Diagnose des Usher-Syndroms basiert auf der beim Patienten beobachteten Kombination aus plötzlicher Taubheit und fortschreitendem Sehverlust.
Tests
Zur Feststellung der Mutation kann ein spezieller Gentest angeordnet werden.
Es wurden elf genetische Loci gefunden, die die Entwicklung des Usher-Syndroms verursachen können, und neun Gene wurden identifiziert, die definitiv die Ursache der Erkrankung sind:
- Typ 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Typ 2: ush2a, VLGR1, WHRN.
- Usher-Syndrom Typ 3: USH3A.
Wissenschaftler des NIDCD haben zusammen mit Kollegen von Universitäten in New York und Israel eine Mutation namens R245X im Pcdh15-Gen identifiziert, die für einen großen Prozentsatz des Usher-Syndroms Typ 1 in der jüdischen Bevölkerung verantwortlich ist.
Um mehr über Labore zu erfahren, die klinische Studien durchführen, besuchen Sie https://www.genetests.org und suchen Sie im Laborverzeichnis nach „Usher-Syndrom“.
Um mehr über bestehende klinische Studien zu erfahren, die genetische Tests auf das Usher-Syndrom beinhalten, besuchen Sie https://www.clinicaltrials.gov und suchen Sie nach „Usher-Syndrom“ oder „Gentests auf Usher-Syndrom“.
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ], [ 30 ]
Instrumentelle Diagnostik
Es gibt verschiedene Methoden der instrumentellen Diagnostik:
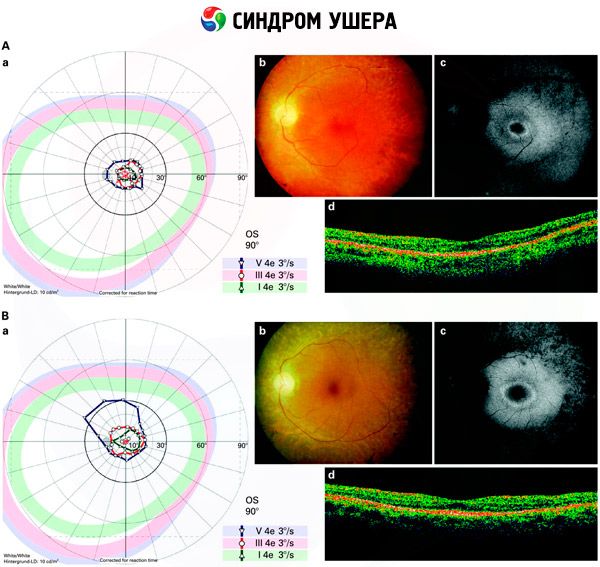
- Untersuchung des Augenhintergrunds, um das Vorhandensein von Pigmentflecken auf der Netzhaut sowie eine Verengung der Netzhautgefäße festzustellen;
- Elektroretinogramm, das die Erkennung beginnender degenerativer Veränderungen der Netzhaut des Auges ermöglicht. Es zeigt das Aussterben elektroradiographischer Bahnen;
- Ein Elektronystagmogramm (ENG) misst unwillkürliche Augenbewegungen, die auf das Vorhandensein eines Ungleichgewichts hinweisen könnten.
- Audiometrie, die verwendet wird, um das Vorhandensein und den Schweregrad einer Taubheit festzustellen.
Differenzialdiagnose
Das Usher-Syndrom muss von einigen ähnlichen Erkrankungen unterschieden werden.
Das Hallgren-Syndrom ist durch angeborenen Hörverlust und fortschreitenden Sehverlust gekennzeichnet (auch Katarakt und Nystagmus entwickeln sich). Weitere Symptome sind Ataxie, psychomotorische Störungen, Psychosen und geistige Behinderung.
Das Alström-Syndrom ist eine Erbkrankheit, bei der die Netzhaut degeneriert und das zentrale Sehvermögen verloren geht. Dieses Syndrom ist mit Fettleibigkeit im Kindesalter verbunden. Gleichzeitig entwickeln sich nach 10 Jahren Diabetes mellitus und Hörverlust.
Röteln bei einer schwangeren Frau im ersten Trimester können verschiedene Entwicklungsstörungen des Kindes verursachen. Zu den Folgen einer solchen Anomalie zählen Hörverlust sowie (oder) Sehstörungen und darüber hinaus verschiedene Entwicklungsstörungen.
Wen kann ich kontaktieren?
Behandlung Usher-Syndrom
Das Usher-Syndrom ist derzeit nicht heilbar. Daher besteht die Therapie in diesem Fall hauptsächlich darin, den Sehverlust zu verlangsamen und den Hörverlust auszugleichen. Mögliche Behandlungsmethoden sind:
- Einnahme von Vitamin A (einige Augenärzte glauben, dass hohe Dosen von Vitamin-A-Palmitat das Fortschreiten der Retinitis pigmentosa verlangsamen, aber nicht stoppen können);
- Implantation spezieller elektronischer Geräte in die Ohren des Patienten (Hörgeräte, Cochlea-Implantate).
Augenärzte empfehlen den meisten Erwachsenen mit häufigen Formen der Retinitis pigmentosa die tägliche Einnahme von 15.000 IE (Internationale Einheiten) Vitamin-A-Palmitat unter Aufsicht. Da Personen mit Usher-Syndrom Typ 1 nicht an der Studie teilnahmen, werden für diese Patientengruppe keine hohen Dosen von Vitamin A empfohlen. Personen, die die Einnahme von Vitamin A in Erwägung ziehen, sollten diese Behandlungsoption mit ihrem Arzt besprechen. Weitere Empfehlungen für diese Behandlungsoption sind:
- Ändern Sie Ihre Ernährung, um Nahrungsmittel mit hohem Vitamin-A-Gehalt einzuschließen.
- Frauen, die eine Schwangerschaft planen, sollten die Einnahme hoher Dosen von Vitamin A drei Monate vor der geplanten Empfängnis abbrechen, da ein erhöhtes Risiko für Geburtsfehler besteht.
- Schwangere sollten aufgrund des erhöhten Risikos von Geburtsfehlern die Einnahme hoher Dosen Vitamin A abbrechen.
Es ist auch wichtig, ein solches Kind an das soziale Leben zu gewöhnen. Dies erfordert die Hilfe von Sonderpädagogen und Psychologen. Falls der Patient einen fortschreitenden Sehverlust erfährt, sollte ihm die Gebärdensprache beigebracht werden.
Prognose
Das Usher-Syndrom hat eine ungünstige Prognose. Bei den meisten Patienten mit dieser Erkrankung jeglicher Art beginnen sich Gesichtsfeld und Sehschärfe im Laufe von 20 bis 30 Jahren zu verschlechtern. In einigen Fällen kommt es zu einem vollständigen beidseitigen Sehverlust. Der Hörverlust, der stets mit Stummheit einhergeht, entwickelt sich sehr schnell zu einem vollständigen beidseitigen Hörverlust.