Facharzt des Artikels

Neue Veröffentlichungen
Xeroderma pigmentosum
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
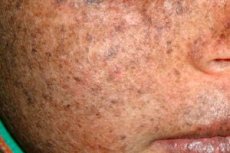
Xeroderma pigmentosum ist eine autosomal-rezessive genetische Störung der DNA-Reparatur. Die Krankheit wird durch Mutationen in Genen verursacht, die an der Reparatur von durch ultraviolette Strahlung und andere toxische Substanzen geschädigter DNA beteiligt sind.
Am häufigsten betrifft diese Krankheit Kinder, die auch "Kinder der Nacht" genannt werden. Häufige Komplikationen der Krankheit sind Basalzellkarzinome und andere bösartige Neubildungen der Haut, metastasiertes malignes Melanom und Plattenepithelkarzinom.
 [ 1 ]
[ 1 ]
Ursachen Xeroderma pigmentosum
Xeroderma pigmentosum ist eine Erbkrankheit, die durch ein autosomales Gen von Eltern und Verwandten übertragen wird und familiären Charakter hat. Lunch (1967) stellte in einer Familie mit sieben Geschwistern bei fünf von ihnen klinische Anzeichen von Xeroderma pigmentosum fest.
Pathogenese
Ein Mangel an UV-Endonuklease-Enzymen in den Zellen des Patienten (in Fibroblasten) oder deren vollständiges Fehlen spielt eine wichtige Rolle bei der Entstehung der Krankheit. Dieses Enzym ist für die Reproduktion von durch ultraviolette Strahlen geschädigter DNA verantwortlich. Anderen Quellen zufolge spielt bei einigen Patienten ein Mangel an DNA-Polymerase-1-Enzym eine wichtige Rolle bei der Entstehung der Krankheit. Die Krankheit entwickelt sich am häufigsten unter dem Einfluss von Strahlen mit einer Wellenlänge von 280–310 nm. Es wird angenommen, dass Pigmentxerodermie durch das Eindringen verschiedener photodynamischer Substanzen in den Körper oder einen Anstieg von Porphyrinen in der menschlichen biologischen Umgebung entsteht.
In den frühen Stadien der Erkrankung treten Hyperkeratose, Atrophie des Epidermisherdes, Ausdünnung der Malpighischen Schicht, eine Zunahme von Melaningranula in der Basalschicht der Zellen und ein chronisches Entzündungsinfiltrat in der oberen Schicht der Dermis (hauptsächlich um die Blutgefäße herum) auf. Anschließend zeigen sich degenerative Veränderungen in Kollagen- und elastischen Fasern, und im Tumorstadium zeigen sich für Hautkrebs charakteristische Anzeichen.
Symptome Xeroderma pigmentosum
Männer und Frauen sind gleichermaßen von dieser Krankheit betroffen. Die Krankheit beginnt in der frühen Kindheit im Frühling oder Sommer. Die Haut des Patienten reagiert besonders empfindlich auf Sonnenlicht. Daher tritt der erste Ausschlag in Form einer Hyperpigmentierung, ähnlich einem Muttermal, auf den dem Sonnenlicht ausgesetzten Hautpartien auf. Der Ausschlag nimmt zu, das Erythem verstärkt sich, wird dunkelbraun, es treten Teleangiektasien, Angiome und Atrophien auf. Anschließend wachsen Papillome und Warzen (hauptsächlich auf der Haut von Gesicht und Hals), es treten Flecken und Geschwüre auf. Infolge der Geschwürvernarbung wird die Nase dünner („Vogelschnabel“), das Augenlid stülpt sich um. Papillome entwickeln sich meist zu bösartigen Tumoren. Pigmentierte Xerodermie tritt in der Regel zusammen mit spinozellulärem Krebs, Melanosarkomen, auf.
Was bedrückt dich?
Bühnen
Im klinischen Verlauf des Xeroderma pigmentosum werden fünf Stadien unterschieden: entzündlich (erythematös), Hyperpigmentierung, Atrophie, Hyperkeratose und bösartige Tumoren.
Im entzündlichen (erythematösen) Stadium treten an sonnenexponierten Hautstellen (Gesicht, Hals, oberer Brustbereich, Arme, Hände) Schwellungen, rote Flecken und manchmal Blasen und Bläschen auf. An sonnenexponierten Stellen tritt fast kein Ausschlag auf.
Bei der Hyperpigmentierung treten anstelle der roten Flecken hellbraune, braune, hyperpigmentierte Flecken auf, die Muttermalen ähneln.
Bei Atrophie trocknet die Haut aus, wird dünner und faltig. Auf der Haut der Lippen und der Nase sind zahlreiche kleine, sternförmige Teleangiektasien und Narben mit glänzender Oberfläche zu beobachten. Aufgrund von Atrophie und Narben entwickeln sich Mikrotomie (Verkleinerung der Mundöffnung), Ektropium, Ausdünnung von Ohren und Nase sowie Atresie der Nasenöffnung und des Mundes. Bei 80–85 % der Patienten sind die Augen betroffen: Konjunktivitis, Keratitis, Schädigung der Hornhaut und Schleimhaut sowie Sehschwäche werden festgestellt. Auf der Haut der Augenlider werden Dyschromie, Teleangiektasien, hyperkeratotische Veränderungen und Tumoren beobachtet.
Bei Hyperkeratose treten in den oben beschriebenen pathologischen Herden Warzentumoren, Papillome, Keratoakanthome, Fibrome, Angiofibriome und andere gutartige Tumoren auf. Daher zählen einige Wissenschaftler die Pigmentxerodermie zur Gruppe der Krebsvorstufen.
10–15 Jahre nach Krankheitsbeginn treten bösartige Hauttumoren (Basaliom, Endotheliom, Angiosarkom) in atrophisch veränderten Herden und Pigmentflecken auf. Die Tumoren zerstören sich innerhalb kurzer Zeit, metastasieren in innere Organe und führen zum Tod. In den inneren Organen und Geweben einiger Patienten werden allgemeine dystrophische Veränderungen beobachtet (Syndaktelien der zweiten und dritten Zehe, Zahndystrophie, vollständiger Haarausfall usw.).
Formen
Die neurologische Form des Xeroderma pigmentosum manifestiert sich in zwei Syndromen.
Das Reed-Syndrom ist durch klinische Manifestationen von Xeroderma pigmentosum und Mikrozephalie, Idiopathie und langsamem Wachstum des Skelettsystems gekennzeichnet. Bei der neurologischen Form der Erkrankung ist die DNA-Reparatur schwierig und die Röntgentherapie führt zu einer Zunahme der wichtigsten pathologischen Herde.
Das De Sinctis-X-Cocchione-Syndrom ist durch folgende klinische Symptome gekennzeichnet:
- Entwicklung klinischer Anzeichen von Xeroderma pigmentosum auf besonders lichtempfindlicher Haut;
- frühes Auftreten bösartiger Tumoren;
- gleichzeitiger Verlauf von spastischer Lähmung, Mikrozephalie und Demenz;
- angeborene Missbildungen und Zwergwuchs;
- Unterentwicklung der Keimdrüsen;
- häufige Fehlgeburten;
- rezessive Vererbung.
Nach Ansicht einiger Dermatologen ist das Santis-Cacchione-Syndrom keine eigenständige Erkrankung, sondern eine schwere und voll ausgeprägte klinische Manifestation des Xeroderma pigmentosum.
Genetische Formen
Arten |
Gen |
Ort |
Beschreibung |
Typ A, I, XPA |
XPA |
9q22.3 |
Xeroderma pigmentosum (XP) Gruppe A – klassische Form |
Typ B, II, XPB |
XPB |
2. Quartal 21 |
XP-Gruppe B |
Typ C, III, XPC |
XPC |
3 Uhr 25 |
XP-Gruppe C |
Typ D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP-Gruppe D oder De-Sanctis-Cacchione-Syndrom |
Typ E, V, XPE |
DDB2 |
11p12-p11 |
XP-Gruppe E |
Typ F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Gruppe F |
Typ G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP-Gruppe G und COFS-Syndrom (cerebro-okulo-fazioskelettales Syndrom) Typ 3 |
Typ V, XPV |
POLH |
6p21.1-p12 |
Variante Xeroderma pigmentosa |
Was muss untersucht werden?
Wie zu prüfen?
Wen kann ich kontaktieren?
Behandlung Xeroderma pigmentosum
Antimalariamittel (Delagyl, Plaquenil, Resoquin usw.) schützen die DNA vor ultravioletten Strahlen, hemmen den Depolymerisationsprozess und reduzieren die Lichtempfindlichkeit (Photosensibilisierung) der Haut. Diese Medikamente wirken entzündungshemmend und hyposensibilisierend. Es wird empfohlen, die allgemeine Therapie zusammen mit Vitamintherapie (B1, B2, PP, B6, B12, A, E), Antihistaminika (Tavegil, Diphenhydramin, Suprastin) und Desensibilisierungsmitteln (Natriumthiosulfat, 10% Calciumchlorid intravenös 10 ml) durchzuführen.
Zur lokalen Behandlung werden Sonnenschutzcremes und -salben verwendet.
Bei der Tumorform der Pigmentxerodermie kommen chirurgische Methoden, flüssiger Stickstoff und Laserstrahlen zum Einsatz. Um sich vor Sonnenlicht zu schützen, müssen Sie lockere Kleidung, Sonnenhüte und Handschuhe tragen.
Prognose
Die meisten Patienten (2/3) sterben vor Erreichen des 15. Lebensjahres. Einige Patienten können 40–50 Jahre alt werden.
[ 20 ]