Facharzt des Artikels

Neue Veröffentlichungen
Kryptogene Epilepsie mit Krampfanfällen bei Erwachsenen
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Nach der bis zum letzten Jahr gültigen internationalen Klassifikation wurde zwischen symptomatischer oder sekundärer Epilepsie, verursacht durch eine Schädigung der Hirnstrukturen, idiopathischer, primärer (eine eigenständige, vermutlich erbliche Erkrankung) und kryptogener Epilepsie unterschieden. Letztere Option bedeutet, dass die moderne Diagnostik keine Ursachen für periodische epileptische Anfälle festgestellt hat und auch keine erbliche Veranlagung nachgewiesen werden kann. Der Begriff „kryptos“ wird aus dem Griechischen als „unbekannter Ursprung“ übersetzt (kryptos – geheim, geheim, genos – erzeugt).
Die Wissenschaft steht nicht still, und vielleicht wird bald der Ursprung periodischer epileptischer Anfälle unbekannter Ätiologie geklärt sein. Experten gehen davon aus, dass kryptogene Epilepsie eine sekundäre symptomatische Erkrankung ist, deren Entstehung mit dem derzeitigen Stand der Diagnostik nicht geklärt werden kann.
 [
[ Epidemiologie
Epilepsie und epileptische Syndrome sind weit verbreitete neurologische Erkrankungen, die oft schwerwiegende Folgen haben. Epileptische Anfälle können bei Menschen jeden Geschlechts und Alters auftreten. Schätzungsweise 5 % der Weltbevölkerung haben im Laufe ihres Lebens mindestens einen Anfall erlitten.
Jedes Jahr wird bei durchschnittlich 30–50 von 100.000 Menschen auf der Erde eine Epilepsie oder ein epileptisches Syndrom diagnostiziert. Am häufigsten treten epileptische Anfälle bei Säuglingen auf (100 bis 233 Fälle pro 100.000 Menschen). Der Höhepunkt der Manifestation liegt in der Perinatalperiode, danach sinkt die Inzidenzrate fast um die Hälfte. Die niedrigsten Raten gibt es bei Menschen im Alter von 25 bis 55 Jahren – etwa 20–30 Fälle pro 100.000 Menschen. Dann steigt die Wahrscheinlichkeit epileptischer Anfälle und ab dem 70. Lebensjahr beträgt die Inzidenzrate 150 Fälle oder mehr pro 100.000 Menschen.
Die Ursachen einer Epilepsie sind in etwa 40 % der Fälle geklärt, sodass eine Erkrankung unbekannter Ätiologie keine Seltenheit ist. Infantile Spasmen (West-Syndrom), eine kryptogene Epilepsie, werden bei vier- bis sechsmonatigen Kindern diagnostiziert. Im Durchschnitt kommt auf 3.200 Säuglinge ein Kind mit dieser Diagnose.
Ursachen kryptogene Epilepsie
Grundlage für die Diagnose einer Epilepsie sind periodische Anfälle, deren Ursache eine ungewöhnlich starke elektrische Entladung ist, die das Ergebnis einer Synchronisation der Aktivität von Gehirnzellen in allen Frequenzbereichen ist, was sich äußerlich im Auftreten sensorischer, motorischer, neurologischer und psychischer Symptome äußert.
Damit ein epileptischer Anfall auftreten kann, müssen sogenannte epileptische Neuronen vorhanden sein, die durch eine Instabilität des Ruhepotentials (der Potentialdifferenz einer nicht erregten Zelle auf der Innen- und Außenseite ihrer Membran) gekennzeichnet sind. Infolgedessen weist das Aktionspotential eines erregten epileptischen Neurons eine deutlich höhere Amplitude, Dauer und Frequenz als normal auf, was zur Entwicklung eines epileptischen Anfalls führt. Es wird angenommen, dass Anfälle bei Menschen mit einer erblichen Veranlagung zu solchen Verschiebungen auftreten, d. h. bei Gruppen epileptischer Neuronen, die ihre Aktivität synchronisieren können. Epileptische Herde bilden sich auch in Hirnarealen mit veränderter Struktur aufgrund von Verletzungen, Infektionen, Intoxikationen und der Entwicklung von Tumoren.
Bei Patienten mit der Diagnose kryptogener Epilepsie lassen sich mit modernen bildgebenden Verfahren keine Anomalien in der Hirnstruktur feststellen, und es gibt auch keine Fälle von Epilepsie in der Familienanamnese. Dennoch erleiden die Patienten relativ häufig epileptische Anfälle unterschiedlicher Art, die schwer zu behandeln sind (möglicherweise gerade weil ihre Ursache unklar ist).
Demnach werden die bekannten Risikofaktoren für das Auftreten epileptischer Anfälle – genetische Veranlagung, Störungen der Gehirnstruktur, Stoffwechselvorgänge im Gehirngewebe, Folgen von Kopfverletzungen oder Infektionsprozesse – bei Untersuchungen und Befragungen nicht erkannt.
Nach der neuen Klassifikation der Epilepsien von 2017 werden sechs ätiologische Kategorien der Krankheit unterschieden. Anstelle der symptomatischen Epilepsie wird nun empfohlen, die Art der Epilepsie anhand der festgestellten Ursache zu bestimmen: strukturell, infektiös, metabolisch, immun oder eine Kombination davon. Idiopathische Epilepsie setzte eine erbliche Veranlagung voraus und wird nun als genetisch bezeichnet. Der Begriff „kryptogen“ wurde durch „unbekannter ätiologischer Faktor“ ersetzt, was die Bedeutung des Wortlauts klarer machte, sich aber nicht änderte.
Die Pathogenese der Epilepsie ist vermutlich wie folgt: Bildung eines epileptischen Fokus, d. h. einer Gemeinschaft von Neuronen mit gestörter Elektrogenese → Bildung epileptischer Systeme im Gehirn (bei übermäßiger Freisetzung exzitatorischer Mediatoren wird eine „Glutamatkaskade“ in Gang gesetzt, die alle neuen Neuronen beeinflusst und zur Bildung neuer Epileptogeneseherde beiträgt) → Bildung pathologischer interneuronaler Verbindungen → Generalisierung der Epilepsie.
Die Haupthypothese zum Mechanismus der Epilepsieentwicklung basiert auf der Annahme, dass der pathologische Prozess durch eine Verletzung des Gleichgewichtszustands zwischen erregenden Neurotransmittern (Glutamat, Aspartat) und solchen, die für Hemmprozesse verantwortlich sind (γ-Aminobuttersäure, Taurin, Glycin, Noradrenalin, Dopamin, Serotonin), ausgelöst wird. Was genau dieses Gleichgewicht in unserem Fall stört, ist unbekannt. Infolgedessen leiden jedoch die Zellmembranen der Neuronen, die Kinetik der Ionenflüsse wird gestört – Ionenpumpen werden inaktiviert und umgekehrt Ionenkanäle aktiviert, die intrazelluläre Konzentration positiv geladener Kalium-, Natrium- und Chlorionen wird gestört. Der pathologische Ionenaustausch durch destrukturierte Membranen führt zu Veränderungen des zerebralen Blutflusses. Eine Funktionsstörung der Glutamatrezeptoren und die Produktion von Autoantikörpern dagegen verursachen epileptische Anfälle. Periodisch wiederkehrende, übermäßig intensive Nervenentladungen, die sich in Form epileptischer Anfälle äußern, führen zu tiefgreifenden Störungen der Stoffwechselvorgänge in den Zellen der Gehirnsubstanz und provozieren die Entstehung des nächsten Anfalls.
Die Besonderheit dieses Prozesses liegt in der Aggressivität der Neuronen des epileptischen Fokus gegenüber den noch unveränderten Hirnarealen, wodurch sie neue Bereiche unterwerfen können. Die Entstehung epileptischer Systeme erfolgt im Zuge der Bildung pathologischer Beziehungen zwischen dem epileptischen Fokus und den strukturellen Komponenten des Gehirns, die den Mechanismus der Epilepsieentwicklung aktivieren können. Zu diesen Strukturen gehören: der Thalamus, das limbische System, die Formatio reticularis des mittleren Hirnstamms. Die Beziehungen zum Kleinhirn, dem Nucleus caudatus des Subkortex und dem anterioren orbitalen Kortex verlangsamen hingegen die Entwicklung von Epilepsie.
Im Verlauf der Krankheitsentwicklung bildet sich ein geschlossenes pathologisches System – das epileptische Gehirn. Seine Entstehung endet mit einer Störung des Zellstoffwechsels und der Interaktion von Neurotransmittern, der Hirndurchblutung, einer zunehmenden Atrophie von Hirngewebe und -gefäßen sowie der Aktivierung spezifischer zerebraler Autoimmunprozesse.
Symptome kryptogene Epilepsie
Die wichtigste klinische Manifestation dieser Erkrankung ist ein epileptischer Anfall. Der Verdacht auf Epilepsie besteht, wenn der Patient mindestens zwei reflexartige (unprovozierte) epileptische Anfälle hatte, deren Ausprägungen sehr unterschiedlich sind. Beispielsweise handelt es sich bei epilepsieähnlichen Anfällen, die durch hohe Temperaturen verursacht werden und nicht im Normalzustand auftreten, nicht um Epilepsie.
Bei Patienten mit kryptogener Epilepsie können Anfälle unterschiedlicher Art und recht häufig auftreten.
Die ersten Anzeichen der Krankheitsentwicklung (vor dem Auftreten vollwertiger epileptischer Anfälle) können unbemerkt bleiben. Zur Risikogruppe gehören Menschen, die in der frühen Kindheit Fieberkrämpfe erlitten haben, mit der Folge einer erhöhten Anfallsbereitschaft. In der Prodromalphase können Schlafstörungen, erhöhte Reizbarkeit und emotionale Labilität auftreten.
Zudem verlaufen Anfälle nicht immer in der klassischen generalisierten Form mit Stürzen, Krämpfen und Bewusstlosigkeit.
Manchmal sind die einzigen frühen Anzeichen Sprachstörungen, der Patient ist bei Bewusstsein, spricht aber nicht und antwortet nicht auf Fragen, oder regelmäßige kurze Ohnmachtsanfälle. Diese dauern nicht lange – ein paar Minuten – und bleiben daher unbemerkt.
Einfache fokale oder partielle (lokale, begrenzte) Anfälle treten leichter auf, deren Manifestationen vom Ort des epileptischen Fokus abhängen. Der Patient verliert während des Anfalls nicht das Bewusstsein.
Bei einem einfachen motorischen Anfall können Tics, Zuckungen der Gliedmaßen, Muskelkrämpfe sowie Drehbewegungen von Rumpf und Kopf auftreten. Der Patient kann unartikulierte Laute von sich geben oder schweigen, Fragen nicht beantworten, schmatzen, sich die Lippen lecken und Kaubewegungen ausführen.
Einfache sensorische Anfälle sind gekennzeichnet durch Parästhesien – Taubheitsgefühl verschiedener Körperteile, ungewöhnliche Geschmacks- oder Geruchsempfindungen, die normalerweise unangenehm sind; Sehstörungen – Lichtblitze, ein Gitter, Flecken vor den Augen, Tunnelblick.
Vegetative Anfälle äußern sich durch plötzlich auftretende Blässe oder Hyperämie der Haut, erhöhte Herzfrequenz, Blutdrucksprünge, Verengung oder Erweiterung der Pupillen, Beschwerden im Magenbereich bis hin zu Schmerzen und Erbrechen.
Psychische Anfälle äußern sich in Derealisation/Depersonalisation und Panikattacken. Sie sind in der Regel Vorläufer komplexer fokaler Anfälle, die bereits mit Bewusstseinsstörungen einhergehen. Der Patient erkennt, dass er einen Anfall hat, kann aber keine Hilfe suchen. Die Ereignisse während des Anfalls werden aus dem Gedächtnis des Patienten gelöscht. Die kognitiven Funktionen der Person sind beeinträchtigt – ein Gefühl der Unwirklichkeit des Geschehens, neue Veränderungen in sich selbst treten auf.
Fokale Anfälle mit anschließender Generalisierung beginnen als einfache (komplexe) Anfälle und entwickeln sich zu generalisierten tonisch-klonischen Paroxysmen. Sie dauern etwa drei Minuten und gehen in einen tiefen Schlaf über.
Generalisierte Anfälle treten in einer schwereren Form auf und werden unterteilt in:
- tonisch-klonisch, tritt in folgender Reihenfolge auf: Der Patient verliert das Bewusstsein, stürzt, sein Körper beugt und streckt sich in einem Bogen, krampfhafte Muskelzuckungen im ganzen Körper beginnen; die Augen des Patienten rollen nach hinten, seine Pupillen sind in diesem Moment erweitert; der Patient schreit, wird blau, weil er für einige Sekunden die Atmung aussetzt, es kommt zu schaumiger Hypersalivation (der Schaum kann aufgrund von Blut darin eine rosa Färbung annehmen, was auf ein Beißen auf die Zunge oder Wange hindeutet); manchmal kommt es zu einer unwillkürlichen Entleerung der Blase;
- myoklonische Anfälle äußern sich in Form von intermittierenden (rhythmischen und arrhythmischen) Muskelzuckungen über mehrere Sekunden im gesamten Körper oder in bestimmten Körperbereichen, die wie Flattern der Gliedmaßen, Hocken, Ballen der Hände zu Fäusten und andere monotone Bewegungen aussehen; das Bewusstsein bleibt insbesondere bei fokalen Anfällen erhalten (dieser Typ wird häufiger in der Kindheit beobachtet);
- Absencen – nicht-konvulsive Anfälle mit kurzfristigem (5–20 Sekunden) Bewusstseinsverlust, der sich darin äußert, dass die Person mit offenen, ausdruckslosen Augen erstarrt und nicht auf Reize reagiert, normalerweise nicht hinfällt, beim Wiedererwachen die unterbrochene Aktivität fortsetzt und sich nicht an den Anfall erinnert;
- atypische Absencen gehen mit Stürzen und unwillkürlicher Blasenentleerung einher, dauern länger an und treten bei schweren Formen der Erkrankung auf, verbunden mit geistiger Behinderung und anderen Symptomen psychischer Störungen;
- atonische Anfälle (akinetische) - der Patient stürzt infolge des Verlusts des Muskeltonus stark (bei fokalen Epilepsien kann es zu einer Atonie einzelner Muskelgruppen kommen: Gesichtsmuskel - Herabhängen des Unterkiefers, Halsmuskel - der Patient sitzt oder steht mit hängendem Kopf), die Dauer des Anfalls beträgt nicht mehr als eine Minute; die Atonie bei Absencen tritt allmählich auf - der Patient sinkt langsam, bei isolierten atonischen Anfällen - stürzt er stark.
In der Zeit nach dem Anfall ist der Patient lethargisch und gehemmt; wird er nicht gestört, schläft er ein (insbesondere nach generalisierten Anfällen).
Epilepsiearten entsprechen den Anfallsarten. Fokale (partielle) Anfälle entwickeln sich in einem lokalen epileptischen Herd, wenn eine ungewöhnlich starke Entladung auf Widerstand in benachbarten Bereichen stößt und erlischt, ohne sich auf andere Teile des Gehirns auszubreiten. In solchen Fällen wird eine kryptogene fokale Epilepsie diagnostiziert.
Der klinische Verlauf der Erkrankung mit begrenztem epileptischen Herd (fokale Form) wird durch dessen Lokalisation bestimmt.
Am häufigsten werden Schäden im Schläfenbereich beobachtet. Der Verlauf dieser Form ist progressiv, Anfälle sind oft gemischter Art und dauern mehrere Minuten. Kryptogene temporale Epilepsie außerhalb von Anfällen äußert sich in Kopfschmerzen, ständigem Schwindel und Übelkeit. Patienten mit dieser Lokalisation klagen über häufiges Wasserlassen. Vor dem Anfall spüren die Patienten eine Aura als Vorboten.
Die Läsion kann im Frontallappen des Gehirns lokalisiert sein. Die Anfälle sind durch Plötzlichkeit ohne prodromale Aura gekennzeichnet. Der Patient zeigt Kopfzuckungen, die Augen rollen unter der Stirn und zur Seite, und automatische, relativ komplexe Gestikulation ist charakteristisch. Der Patient kann das Bewusstsein verlieren, stürzen und tonisch-klonische Muskelkrämpfe im gesamten Körper entwickeln. Bei dieser Lokalisation kommt es zu einer Reihe von kurzzeitigen Anfällen, manchmal mit Übergang in einen generalisierten und/oder Status epilepticus. Sie können nicht nur tagsüber, sondern auch nachts auftreten. Eine sich entwickelnde kryptogene Frontalepilepsie verursacht psychische Störungen (gewalttätiges Denken, Derealisation) und das autonome Nervensystem.
Sensorische Anfälle (Gefühl von warmer Luft, die über die Haut streicht, leichte Berührungen) verbunden mit krampfhaftem Zucken von Körperteilen, Sprach- und Bewegungsstörungen, Atonie, begleitet von Harninkontinenz.
Die Lokalisierung des epileptischen Herdes im orbital-frontalen Bereich äußert sich in Geruchshalluzinationen, Hypersalivation, epigastrischen Beschwerden sowie Sprachstörungen, Husten und Kehlkopfödemen.
Wenn sich die elektrische Hyperaktivität über alle Teile des Gehirns ausbreitet, entwickelt sich ein generalisierter Anfall. In diesem Fall wird eine kryptogene generalisierte Epilepsie diagnostiziert. Die Anfälle sind in diesem Fall durch Intensität und Bewusstlosigkeit gekennzeichnet und enden mit einem längeren Schlaf. Nach dem Aufwachen klagen die Patienten über Kopfschmerzen, Sehstörungen, Müdigkeit und Leeregefühl.
Es gibt auch eine kombinierte (wenn sowohl fokale als auch generalisierte Anfälle auftreten) und unbekannte Art der Epilepsie.
Kryptogene Epilepsie bei Erwachsenen gilt nicht ohne Grund als sekundär mit einem unbekannten ätiologischen Faktor. Sie ist durch plötzlich auftretende Anfälle gekennzeichnet. Abgesehen von den klinischen Symptomen weisen Epileptiker eine instabile Psyche, ein explosives Temperament und eine Tendenz zur Aggression auf. Die Krankheit beginnt meist mit fokalen Manifestationen. Im weiteren Verlauf breiten sich die Läsionen auf andere Hirnareale aus; das fortgeschrittene Stadium ist durch persönliche Degradation und ausgeprägte psychische Abweichungen gekennzeichnet, und der Patient wird sozial unangepasst.
Die Krankheit verläuft progressiv und die klinischen Symptome der Epilepsie verändern sich je nach Entwicklungsstadium der Epilepsie (Ausprägungsgrad des epileptischen Herdes).
Komplikationen und Konsequenzen
Selbst bei leichter fokaler Epilepsie mit isolierten, seltenen Anfällen werden Nervenfasern geschädigt. Die Krankheit verläuft schleichend: Ein Anfall erhöht die Wahrscheinlichkeit des nächsten und der Bereich der Hirnschädigung weitet sich aus.
Generalisierte, häufige Anfälle haben eine zerstörerische Wirkung auf das Hirngewebe und können sich zu einem Status epilepticus mit hoher Wahrscheinlichkeit eines tödlichen Ausgangs entwickeln. Es besteht auch das Risiko eines Hirnödems.
Komplikationen und Folgen hängen vom Grad der Schädigung der Gehirnstrukturen, der Schwere und Häufigkeit der Anfälle, Begleiterkrankungen, dem Vorhandensein von schlechten Gewohnheiten, dem Alter, der Angemessenheit der gewählten Behandlungstaktik und Rehabilitationsmaßnahmen sowie der verantwortungsvollen Einstellung des Patienten zur Behandlung selbst ab.
In jedem Alter können bei Stürzen Verletzungen unterschiedlicher Schwere auftreten. Hypersalivation und Würgereiz während eines Anfalls erhöhen das Risiko, dass flüssige Substanzen in die Atemwege gelangen und eine Aspirationspneumonie entwickeln.
In der Kindheit kommt es zu Instabilitäten in der geistigen und körperlichen Entwicklung. Die kognitiven Fähigkeiten leiden häufig darunter.
Der psychoemotionale Zustand ist instabil – Kinder sind reizbar, launisch, oft aggressiv oder apathisch, es fehlt ihnen an Selbstbeherrschung und sie passen sich schlecht an die Gruppe an.
Bei Erwachsenen werden diese Risiken durch Verletzungen bei Arbeiten verstärkt, die erhöhte Aufmerksamkeit erfordern. Bei Anfällen kommt es zu Bissen in die Zunge oder Wange.
Bei Epileptikern besteht ein erhöhtes Risiko für Depressionen, psychische Störungen und soziale Fehlanpassung. Menschen mit Epilepsie sind in ihrer körperlichen Aktivität und Berufswahl eingeschränkt.
Diagnose kryptogene Epilepsie
Bei der Diagnose einer Epilepsie werden viele verschiedene Methoden eingesetzt, um diese Krankheit von anderen neurologischen Erkrankungen zu unterscheiden.
Zunächst muss der Arzt die Beschwerden des Patienten oder seiner Eltern, falls es sich um ein Kind handelt, anhören. Es wird eine Anamnese der Krankheit erstellt – Details zur Manifestation, die Besonderheiten des Verlaufs (Häufigkeit von Anfällen, Ohnmacht, Art der Krämpfe und andere Nuancen), die Dauer der Krankheit, das Vorhandensein ähnlicher Erkrankungen bei den Angehörigen des Patienten. Diese Untersuchung ermöglicht es uns, die Art der Epilepsie und die Lokalisation des epileptischen Fokus zu vermuten.
Blut- und Urintests werden verordnet, um den allgemeinen Zustand des Körpers zu beurteilen, das Vorhandensein von Faktoren wie Infektionen, Vergiftungen, biochemischen Störungen festzustellen und das Vorhandensein genetischer Mutationen beim Patienten festzustellen.
Neuropsychologische Tests dienen der Beurteilung der kognitiven Fähigkeiten und des emotionalen Zustands. Regelmäßige Überwachungen ermöglichen die Beurteilung der Auswirkungen der Krankheit auf Nervensystem und Psyche sowie die Bestimmung der Epilepsieart.
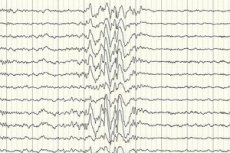
In erster Linie handelt es sich jedoch um eine instrumentelle Diagnostik, mit deren Hilfe die Intensität der elektrischen Aktivität der Gehirnregionen (Elektroenzephalographie), das Vorhandensein von Gefäßfehlbildungen, Neoplasien, Stoffwechselstörungen usw. in ihren Regionen beurteilt werden kann.
Die Elektroenzephalographie (EEG) ist die wichtigste Diagnosemethode, da sie Abweichungen von der Norm in der Gehirnwellenintensität auch außerhalb eines Anfalls zeigt – erhöhte Anfallsbereitschaft bestimmter Bereiche oder des gesamten Gehirns. Das EEG-Muster einer kryptogenen partiellen Epilepsie ist eine Spike-Wave- oder anhaltende langsame Wellenaktivität in bestimmten Teilen des Gehirns. Mithilfe dieser Untersuchung kann die Art der Epilepsie basierend auf der Spezifität des Elektroenzephalogramms bestimmt werden. Beispielsweise ist das West-Syndrom durch unregelmäßige, praktisch unsynchronisierte arrhythmische langsame Wellen mit abnorm hoher Amplitude und Spike-Entladungen gekennzeichnet. In den meisten Fällen des Lennox-Gastaut-Syndroms zeigt das Elektroenzephalogramm im Wachzustand eine unregelmäßige generalisierte langsame Spike-Wave-Aktivität mit einer Frequenz von 1,5–2,5 Hz, oft mit Amplitudenasymmetrie. Während der Nachtruhe ist dieses Syndrom durch die Registrierung schneller rhythmischer Entladungen mit einer Frequenz von etwa 10 Hz gekennzeichnet.
Bei kryptogener Epilepsie ist dies die einzige Möglichkeit, ihr Vorliegen zu bestätigen. Es gibt jedoch Fälle, in denen das EEG selbst unmittelbar nach einem Anfall keine Veränderungen in der Form der Gehirnströme registriert. Dies kann ein Zeichen dafür sein, dass Veränderungen der elektrischen Aktivität in den tiefen Hirnstrukturen auftreten. Veränderungen im EEG können auch bei Patienten ohne Epilepsie auftreten.
Es werden zwangsläufig moderne Methoden der Neurovisualisierung eingesetzt – Computer-, Resonanz- und Positronen-Emissions-Tomographie. Diese instrumentelle Diagnostik ermöglicht es, Veränderungen in der Struktur der Hirnsubstanz aufgrund von Verletzungen, angeborenen Anomalien, Krankheiten, Intoxikationen zu beurteilen und Neoplasien usw. zu erkennen. Die Positronen-Emissions-Tomographie, auch funktionelle MRT genannt, hilft, nicht nur strukturelle, sondern auch funktionelle Störungen zu erkennen.
Tiefere Herde abnormer elektrischer Aktivität können durch Einzelphotonen-Emissionscomputertomographie erkannt werden, und durch Resonanzspektroskopie können Störungen biochemischer Prozesse im Hirngewebe erkannt werden.
Eine experimentelle und wenig verbreitete Diagnosemethode ist die Magnetenzephalographie. Dabei werden die von Neuronen im Gehirn ausgesendeten magnetischen Wellen aufgezeichnet. Sie ermöglicht die Untersuchung tiefster Strukturen des Gehirns, die für die Elektroenzephalographie unzugänglich sind.
Differenzialdiagnose
Die Differentialdiagnose erfolgt nach Durchführung umfassender Untersuchungen. Die Diagnose einer kryptogenen Epilepsie wird durch den Ausschluss anderer im Rahmen der Diagnostik identifizierter Arten und Ursachen epileptischer Anfälle sowie einer erblichen Veranlagung gestellt.
Nicht alle medizinischen Einrichtungen verfügen über das gleiche diagnostische Potenzial, daher erfordert eine solche Diagnose weitere diagnostische Forschung auf einem höheren Niveau.
Behandlung kryptogene Epilepsie
Es gibt keine einheitliche Methode zur Behandlung von Epilepsie. Es wurden jedoch klare Standards entwickelt, die befolgt werden, um die Qualität der Behandlung und das Leben der Patienten zu verbessern.
Verhütung
Da die Ursachen dieser besonderen Form der Epilepsie noch nicht geklärt sind, stehen vorbeugende Maßnahmen im Vordergrund. Ein gesunder Lebensstil – keine schlechten Angewohnheiten, gute Ernährung und körperliche Aktivität – sorgt für eine gute Immunität und beugt der Entstehung von Infektionen vor.
Wenn Sie Ihrer Gesundheit große Aufmerksamkeit schenken und Krankheiten und Verletzungen rechtzeitig untersuchen und behandeln, erhöht sich auch die Wahrscheinlichkeit, diese Krankheit zu vermeiden.
Prognose
Kryptogene Epilepsie manifestiert sich in jedem Alter und weist keinen spezifischen Symptomkomplex auf, sondern äußert sich sehr vielfältig – verschiedene Anfallsarten und Syndrome sind möglich. Bislang gibt es keine einheitliche Methode zur vollständigen Heilung von Epilepsie, aber eine antiepileptische Behandlung hilft in 60-80 % der Fälle aller Krankheitsarten.
Im Durchschnitt dauert die Krankheit zehn Jahre, danach können die Anfälle aufhören. 20 bis 40 % der Patienten leiden jedoch ihr ganzes Leben lang an Epilepsie. Etwa ein Drittel aller Patienten mit Epilepsie jeglicher Art stirbt an den damit verbundenen Ursachen.
Beispielsweise haben kryptogene Formen des West-Syndroms eine ungünstige Prognose. In den meisten Fällen entwickeln sie sich zum Lennox-Gastaut-Syndrom, dessen leichte Formen medikamentös behandelt werden können, während generalisierte Formen mit häufigen und schweren Anfällen lebenslang bestehen bleiben und mit einer schweren geistigen Beeinträchtigung einhergehen können.
Generell hängt die Prognose stark vom Zeitpunkt des Behandlungsbeginns ab; wird im Frühstadium mit der Behandlung begonnen, ist die Prognose günstiger.
Epilepsie kann zu lebenslanger Behinderung führen. Entwickelt eine Person infolge der Krankheit eine anhaltende Gesundheitsstörung, die zu einer Einschränkung der Lebensaktivitäten führt, wird dies durch eine ärztliche und soziale Untersuchung festgestellt. Sie entscheidet auch über die Zuordnung zu einer bestimmten Behinderungsgruppe. Wenden Sie sich hierzu zunächst an Ihren behandelnden Arzt, der den Patienten der Kommission vorstellt.