Facharzt des Artikels

Neue Veröffentlichungen
Kryptogene Epilepsie bei Kindern
Zuletzt überprüft: 07.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
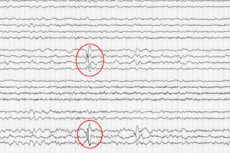
Dieser medizinische Bericht stellt keine endgültige Diagnose dar; die Symptome können sich mit zunehmendem Alter verändern und eine bekannte Form annehmen oder sich zurückbilden.
Die kryptogene Epilepsie umfasst die bekanntesten epileptischen Syndrome, die sich im Kindesalter manifestieren.
- Das West-Syndrom (infantile oder infantile Spasmen) tritt erstmals im Säuglingsalter auf. Diese Diagnose liegt bereits bei Babys im Alter von vier bis sechs Monaten vor, männliche Säuglinge sind anfälliger für diese Krankheit. Das Syndrom ist durch häufige Anfälle gekennzeichnet, die nicht auf eine medikamentöse Therapie ansprechen. Ein Elektroenzephalogramm zeigt eine chaotische zerebrale Hyperaktivität, und die frühe psychomotorische Entwicklung des Kindes ist beeinträchtigt.
- Das Lennox-Gastaut-Syndrom manifestiert sich bei älteren Kindern mit folgenden Symptomen: Kinder stürzen plötzlich (Atonie), bleiben dabei bei Bewusstsein und werden manchmal für einen Moment ohnmächtig. Sie haben normalerweise keine Krämpfe. Der Anfall vergeht sehr schnell und das Kind ist bereits wieder auf den Beinen.
Bei diesem Syndrom können Sturzanfälle mit asthenisch-astatischen, myoklonisch-statischen und tonischen Anfällen sowie atypischen Absencen auftreten. Am häufigsten wird das Lennox-Gastaut-Syndrom bei vier- bis sechsjährigen Kindern diagnostiziert, es kann jedoch auch bei zwei- bis dreijährigen Kindern und bei Achtjährigen auftreten.
In etwa der Hälfte der Fälle wurde das West-Syndrom im Säuglingsalter diagnostiziert; in anderen Fällen manifestierte sich das Lennox-Gastaut-Syndrom nach dem zweiten Lebensjahr als eigenständige Erkrankung.
Tonische Anfälle treten zu jeder Tageszeit auf und sind vielfältig. Sie sind gekennzeichnet durch: plötzliches Beugen von Hals und Körper; die Arme des Patienten sind meist halb gebeugt angehoben. Es sind Kontraktionen der Gesichtsmuskulatur erkennbar, der Patient verdreht die Augen, sein Gesicht wird rot und seine Atmung setzt aus.
Das Lennox-Gastaut-Syndrom ist auch durch atypische Absencen gekennzeichnet, die sich durch eine Vielzahl von Symptomen auszeichnen. Gleichzeitig kann ein gewisses Bewusstsein sowie eine eingeschränkte motorische und sprachliche Aktivität vorhanden sein. Häufig sind auch Hypersalivation, vollständiges oder teilweises Fehlen von Mimik, Myoklonus des Mundes und der Augenlider sowie verschiedene atonische Phänomene: Der Kopf fällt hilflos auf die Brust, der Mund öffnet sich leicht. Atypische Absencen treten häufig vor dem Hintergrund eines verminderten Muskeltonus auf, der wie eine „Erschlaffung“ des Körpers aussieht und meist von oben – den Gesichts- und Nackenmuskeln – ausgeht. Ein intellektuelles Defizit bei der kryptogenen Variante dieses Syndroms entwickelt sich unmittelbar nach Beginn der Anfälle.
- Generalisierte Krampfanfälle manifestieren sich als myoklonisch-astatische Form der kryptogenen Epilepsie. Sie treten meist im Alter zwischen zehn Monaten und fünf Jahren auf. Am häufigsten manifestieren sich solche Anfälle im Alter von ein bis drei Jahren, näher am fünften Lebensjahr treten myoklonische und myoklonisch-astatische Anfälle auf. Äußerlich äußert sich der Anfall in krampfartigen, sehr schnellen Bewegungen der Arme und Beine, verbunden mit häufigem Kopfnicken und einem leichten, pulsierenden Krampf, der den Körper durchläuft. Der Patient fällt, als hätte er einen Schlag auf die Knie bekommen. Die Anfälle treten meist morgens nach dem Aufstehen auf.
Auch die Symptome epileptischer Anfälle unterscheiden sich je nach Altersgruppe der Patienten.
Im Säuglingsalter können Krampfanfälle mit Fieberkrämpfen oder Hyperaktivität verwechselt werden. Das Kind kann Fieber entwickeln, weinerlich und reizbar werden, und die Krampfanfälle beginnen meist auf einer Körperseite und wandern allmählich auf die andere. Schaumiger Hypersalivation sowie Schlaf- und Appetitstörungen werden in der Regel nicht beobachtet.
In der frühen Kindheit treten normalerweise keine Krämpfe auf. Anfälle treten in Form einer Loslösung von der umgebenden Realität auf. Das Kind erstarrt mit einem konzentrierten Blick "in sich selbst", reagiert nicht auf die Behandlung.
Schulkinder, insbesondere Jungen, klagen über Missempfindungen der Haut und Schleimhaut der Mundhöhle und des Kehlkopfes. Der Unterkiefer verschiebt sich zur Seite, die Zähne klappern, die Zunge zittert, es kommt zu vermehrtem Speichelfluss und undeutlicher Sprache. Die Anfälle beginnen oft im Schlaf.
Jugendliche erleben Muskelkrämpfe im ganzen Körper, Verspannungen im Rumpf und in den Gliedmaßen vor dem Hintergrund einer Ohnmacht sowie unwillkürliche Darm- und Blasenentleerung. Während eines Anfalls drehen Patienten oft den Kopf und werfen ihn zur einen oder anderen Seite zurück.
 [
[