Facharzt des Artikels

Neue Veröffentlichungen
Phenylpyruvin-Oligophrenie oder Phenylketonurie
Zuletzt überprüft: 05.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
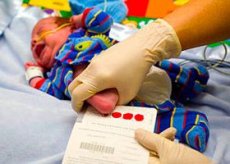
Laut Statistik wird bei einem von 10.000 bis 15.000 Neugeborenen eine genetisch bedingte neurokognitive Pathologie diagnostiziert – die Phenylpyruvat-Oligophrenie, die aufgrund einer angeborenen Stoffwechselstörung der essentiellen Aminosäure Phenylalanin entsteht.
Die Krankheit wurde erstmals in den 1930er Jahren in Norwegen vom Arzt Ivar Foelling entdeckt und als Hyperphenylalaninämie bezeichnet. Heute wird die Krankheit allgemein als Phenylketonurie bezeichnet und hat den ICD-10-Code E70.0.
 [
[ Ursachen der Phenylpyruvat-Oligophrenie
Genetische Ursachen der Phenylbrenztrauben-Oligophrenie sind die rezessive Vererbung durch ein Kind von Eltern, die Träger zweier mutierter Allele des Enzyms Phenylalaninhydroxylase (Phenylalanin-4-Monooxygenase) sind. Infolgedessen fehlt dem Kind dieses Enzym, das für den Abbau von Phenylalanin (proteinogene α-Amino-β-phenylpropionsäure) zu Tyrosin [2-Amino-3-(4-hydroxyphenyl)propansäure] notwendig ist, ganz oder teilweise.
Genetiker haben festgestellt, dass die Pathogenese dieser Krankheit mit mehr als 500 verschiedenen homozygoten oder heterozygoten Strukturmutationen auf Chromosom 12 – in den Genen des Leberenzyms Phenylalaninhydroxylase 12q23.2 oder 12q22-q24.1 – zusammenhängt. Die Genmutation führt dazu, dass der Spiegel dieses Enzyms, das für die Oxidation des in proteinhaltigen Lebensmitteln enthaltenen Phenylalanins notwendig ist, um das Vierfache oder mehr abnimmt.
Dies führt einerseits zu einer Ansammlung von überschüssigem Phenylalanin im Blut, das für die Entwicklung des Gehirns toxisch ist. In diesem Fall wird Phenylalanin, das nicht in Tyrosin umgewandelt werden kann, durch Desaminierung zu Phenylbrenztraubensäure (2-Oxo-3-phenylpropionsäure oder Phenylpyruvat) abgebaut. Diese Säure wird wiederum in Phenylessigsäure (Phenylacetat) und Phenylmilchsäure (Phenyllactat) umgewandelt, die sich negativ auf die Zellen des Gehirns und des zentralen Nervensystems auswirken.
Andererseits führen die Hauptursachen der Phenylbrenztrauben-Oligophrenie (angeborener Mangel an Phenylalaninhydroxylase und dessen Folge – ein Überschuss an Phenylalanin) zu einer Verringerung der Spiegel anderer Aminosäuren im Gehirn (Tryptophan, Threonin, Methionin, Valin, Isoleucin, Leucin), wodurch der Prozess der Biosynthese der Neurotransmitter, die Nervenimpulse übertragen – Dopamin und Noradrenalin – völlig gestört wird und eine fortschreitende Beeinträchtigung der geistigen und körperlichen Entwicklung bei Kindern verursacht wird.
Symptome der Phenylpyruvat-Oligophrenie
Ein Überschuss an Phenylalanin im Körper macht sich bei Säuglingen bemerkbar. Die ersten Anzeichen sind: Lethargie und Schläfrigkeit, Probleme beim Saugen, Krämpfe, Erbrechen, ekzematöse Läsionen der Epidermis, muffiger (mausartiger) Geruch von Körper, Urin und Atem (aufgrund des erhöhten Gehalts an Phenylacetat, das zu Phenylketonen oxidiert wird).
Die folgenden typischen Symptome einer Phenylbrenztrauben-Oligophrenie werden beobachtet: Wachstumsverzögerung, Muskelhyperkinese, Zittern oder verminderter Muskeltonus, psychotische Anfälle, begleitet von grundloser Reizbarkeit und Wut, verminderte intellektuelle Fähigkeiten oder geistige Behinderung.
Kinder mit Phenylketonurie weisen zudem eine Hypopigmentierung auf – eine deutlich hellere und dünnere Haut-, Haar- und Augenfarbe im Vergleich zu ihren Eltern. Die unmittelbare Ursache dieses Symptoms ist ein Mangel an Tyrosin, das bei Oxidation in den Melanozyten der Epidermis das Pigment Melanin bildet.
Wie Spezialisten der Kinderpsychiatrie anmerken, hängt das klinische Bild der Phenylpyruvat-Oligophrenie vom Grad der Schädigung der Gehirnzellen ab, und ein betroffenes Baby kann in den ersten Lebensmonaten normal erscheinen. Wurde die Krankheit nicht unmittelbar nach der Geburt erkannt und das Kind über längere Zeit nicht behandelt, hat dies Folgen: irreversible Hirnschäden und deren Komplikationen in Form schwerer oder tiefgreifender geistiger Behinderung (Schwachsinn oder Idiotie).
Und dann können folgende Symptome einer Phenylbrenztrauben-Oligophrenie auftreten: Kleinwuchs, kraniofaziale Anomalien (Mikrozephalie, vorstehender Oberkiefer, weit auseinanderstehende Zähne, gestörte Zahnschmelzentwicklung), erhöhter Muskeltonus und erhöhte Sehnenreflexe, Hyperaktivität, schlechte Bewegungskoordination und ungelenker Gang, erhöhte Reizbarkeit und andere Verhaltens- und neurologische Probleme bis hin zu psychischen Störungen.
Diagnose von Phenylpyruvat-Oligophrenie
Eine frühzeitige Diagnose der Phenylpyruvat-Oligophrenie und eine sofortige Behandlung sind äußerst wichtig. Dadurch wird das Risiko einer geistigen Behinderung von 80–90 % auf 6–8 % gesenkt (laut CLIMB – National Information Centre for Metabolic Diseases, Großbritannien).
Zu diesem Zweck sollte bei Säuglingen am dritten bis vierten Tag nach der Geburt (in Ausnahmefällen zwischen dem 5. und 8. Tag) ein Neugeborenenscreening auf Phenylketonurie mittels einer mikrobiologischen Blutuntersuchung auf den Phenylalaningehalt – dem sogenannten Guthrie-Test – durchgeführt werden.
Biochemische Urintests auf Phenylpyruvat zeigen auch bei Neugeborenen eine Phenylketonurie an, dieser Test sollte jedoch erst 10-12 Tage nach der Geburt durchgeführt werden. Daher ist der internationale diagnostische Standard die Bestimmung der Phenylalaninkonzentration im Blutplasma.
Moderne instrumentelle Diagnostik auf Basis neuer Technologien wie der Tandem-Massenspektrometrie ermöglicht die Erkennung vieler angeborener Stoffwechselstörungen, die zu geistigen Behinderungen führen. Dies gewährleistet die Differentialdiagnostik genetisch bedingter Stoffwechselerkrankungen, darunter Galaktosämie, Ketonurie (Störungen des Leucin-, Isoleucin- und Valinstoffwechsels), Homocystinurie, Glutarazidurie, Isovalerianazidämie, Sichelzellenanämie, Tyrosinämie, angeborene Hypothyreose usw.
Wen kann ich kontaktieren?
Behandlung von Phenylpyruvat-Oligophrenie
Eine Diättherapie ist praktisch die einzige wirksame Behandlung der Phenylpyruvat-Oligophrenie, die eine normale Entwicklung des Gehirns ermöglicht. Zu Beginn der Behandlung bei Kindern über drei oder vier Jahren beseitigt eine Diättherapie jedoch nicht die bereits aufgetretenen Symptome der Pathologie.
Säuglinge mit Phenylalaninhydroxylasemangel können keine Muttermilch zu sich nehmen und es gibt spezielle Säuglingsnahrungen ohne Phenylalanin für sie – Lofenalac, Afenilak, Aminogran, Milupa pku1 (pku2 und pku3), Periflex Infanta, XP Maxamum.
Die Diät schließt den Verzehr von Fleisch, Fisch, Geflügel, Milch, Eiern, Käse, Hüttenkäse, Eiscreme, Hülsenfrüchten, Nüssen und vielen anderen proteinhaltigen Produkten aus. Kaseinhydrolysat wird als Lebensmittelersatz für verbotene Produkte verwendet, um das Proteingleichgewicht im Körper aufrechtzuerhalten - eine Mischung von Aminosäuren ohne Phenylalanin, die aus Milchprotein gewonnen werden (Medikamente Berlofan oder Ipofenat).
Während frühere Ärzte glaubten, dass die Einnahme von Phenylalanin-freien Mischungen und die Einhaltung einer speziellen Diät bis zu 10 Jahre (also bis zum Abschluss der Myelinisierungsprozesse im Gehirn) fortgesetzt werden sollten, ist heute bekannt, dass ein Absetzen der Behandlung zu einem intensiveren Verlauf der Phenylpyruvat-Oligophrenie und irreversiblen Folgen führen kann. Während Patienten, die diätetische Einschränkungen einhalten, keine Symptome zeigen.
Ein Medikament zur Behandlung der Phenylpyruvat-Oligophrenie (Phenylketonurie) ist heute das Medikament Kuvan auf Basis von Sapropterindihydrochlorid, einem synthetischen Ersatz für Tetrahydrobiopterin (BH4), den Cofaktor des Enzyms Phenylalaninhydroxylase. Kuvan-Tabletten werden in Wasser aufgelöst einmal täglich eingenommen – morgens zu den Mahlzeiten. Die Dosis wird individuell berechnet – entsprechend dem Körpergewicht des Patienten (10 mg pro Kilogramm). Zu den Nebenwirkungen des Medikaments zählen wässriger Nasenausfluss, verstopfte Nase, Kopfschmerzen, Kehlkopfschmerzen, Erbrechen, Durchfall und gelegentlich Bauchschmerzen. Die Einnahme dieses Medikaments hebt die Anti-Phenylalanin-Diät nicht auf. In der Gebrauchsanweisung des Medikaments wird darauf hingewiesen, dass seine Anwendung bei Kindern unter vier Jahren nicht untersucht wurde.
In diesem Fall erfolgt die Behandlung der Phenylbrenztrauben-Oligophrenie unter ständiger Überwachung des Phenylalaninspiegels im Blut.
Phenylpyruvat-Oligophrenie oder Phenylketonurie ist eine Erbkrankheit und kann nicht verhindert werden. Bei Kinderwunsch ist jedoch eine Prävention möglich – ein Enzymbluttest und ein Gentest, um festzustellen, ob zukünftige Eltern Träger des mutierten Gens sind. Blutphenylalanintests können auch während der Schwangerschaft durchgeführt werden.
Wie das Journal of Inherited Metabolic Disease schreibt, „beträgt die Prognose für Patienten mit dieser Erkrankung unbehandelt eine maximale Lebenserwartung von bis zu 30 Jahren im Status eines Behinderten mit schwerer Beeinträchtigung der Gehirnfunktion (da der Phenylalaninspiegel im Blut mit der Zeit ansteigt) und mit Behandlung – bis ins hohe Alter, mit höherer Bildung und einer erfolgreichen Karriere.“