Facharzt des Artikels

Neue Veröffentlichungen
Parietales Meningeom
Zuletzt überprüft: 07.06.2024

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
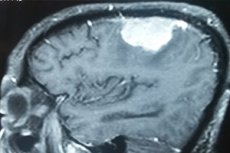
Das parietale Meningiom oder das parietale Meningiom ist ein Tumor, der aus modifizierten Meningothelzellen der mittleren Dura-Mater stammt, die an die innere Schicht der Dura Mater über den parietalen Lappen (lobus parietalis) des Gehirnrortens gebunden ist. Die meisten Tumoren dieses Typs (80-90%) sind gutartig.
Epidemiologie
Meningiome machen 37,6% aller primären ZNS-Tumoren und 53,3% der nichtmalignen intrakraniellen Tumoren aus; In weniger als 10% der Fälle treten mehrere Meningiome auf. Sie treten am häufigsten bei Erwachsenen 40 bis 60 Jahren an und werden bei Kindern selten nachgewiesen. Diese Neoplasien sind bei Frauen dreimal häufiger als bei Männern.
Meningiome der Klasse II machen bis zu 5-7% der Fälle aus, während Meningiome der Grad III 1-2% ausmachen.
Das parietale Meningiom ist eine ziemlich seltene Diagnose.
Ursachen Parietale Meningeome
Meningiom gilt als der häufigste primäre intrakranielle Tumor, es wird durch pathologisch bewachsene Meningothelzellen des Spinnennetzes (Arachnoidea mater Encephali) des Gehirns gebildet. [1]
Typischerweise treten Meningiome spontan auf, was bedeutet, dass die Ursachen unbekannt sind.
Es wird angenommen Defekte (Mutationen, Aberrationen, Spleißen, Amplifikation oder Verlust) von Genen, die die Rate der Zellteilung (aufgrund von Proteinwachstumsfaktoren) und den Prozess der Zellapoptose regulieren.
Beispielsweise führt eine genetische Störung wie den Verlust von Chromosom 22Q zu einem familiären Syndrom, neurofibromatose typ 2, was nicht nur für viele Fälle von Meningiomen, sondern auch für eine erhöhte Inzidenz anderer Hirntumoren berücksichtigt wird.
Meningiome sind in drei Klassen unterteilt: gutartig (Grad I), atypisch (Grad II) und anaplastisch oder bösartig (Grad III). Histologische Meningiome sind ebenfalls unterschieden: fibröses, pammomatisches, gemischtes usw.
Risikofaktoren
Bisher ist der einzige nachgewiesene Faktor, der das Risiko einer Meningiome erhöht, die Exposition gegenüber ionisierender Strahlung (Strahlung) in die Kopfregion (insbesondere in der Kindheit).
Sie fanden auch einen Zusammenhang zwischen der Entwicklung dieser Art von Tumor und Fettleibigkeit, die die Forscher auf eine erhöhte Signalübertragung von Insulin und Insulin-ähnlichen Wachstumsfaktor (IGF-1) zurückführten, die die Zellapoptose hemmen und das Tumorwachstum stimulieren.
Einige Forscher haben ein erhöhtes Meningiom in Menschen festgestellt, die beruflich mit dem Einsatz von Pestiziden und Herbiziden assoziiert sind.
Pathogenese
Die Meningothelzellen des Spinnennetzes stammen aus Keimgewebe (Mesenchym); Sie bilden dichte interzelluläre Kontakte (Desmoses) und erzeugen zwei Barrieren gleichzeitig: zwischen Cerebrospinalflüssigkeit und nervöses Gewebe sowie zwischen dem Alkohol und dem Kreislauf.
Diese Zellen werden durch die Spinne und die weiche Gehirnmembran (PIA Mater Encephali) sowie die Spinnensepta und die Krawatten, die den Subarachnoidalraum überqueren, ein Raum mit einer Flüssigkeit zwischen Spinnen und weichen Cerebralmembranen gefüllt.
Der molekulare Mechanismus einer erhöhten Proliferation von umhüllten Meningothelzellen und die Pathogenese der sporadischen Meningiombildung sind kaum bekannt.
Ein gutartiger Tumor (Grad-I-Meningiom) mit einer ausgeprägten abgerundeten Form und Basis, die Zellen, die es bilden, wächst nicht in das umgebende Gehirngewebe, wächst normalerweise im Schädel und setzt fokale Druck auf benachbarte oder untere Gehirngewebe aus. Tumoren können auch nach außen wachsen und die Schädelverdickung (Hyperostose) verursachen. Bei anaplastischen Meningiomen kann das Wachstum invasiv sein (Ausbreitung auf das Gehirngewebe).
Histologische Studien haben gezeigt, dass viele Tumoren dieser Art einen Bereich mit der höchsten proliferativen Aktivität haben. Und es gibt eine Hypothese, nach der Meningiome aus einem bestimmten neoplastischen transformierten Zellklon gebildet werden, der sich durch die Gehirnmembranen ausbreitet.
Symptome Parietale Meningeome
Es ist schwierig, die ersten Anzeichen eines Meningioms der parietalen Region zu bemerken, da Kopfschmerzen ein unspezifisches Symptom sind und bei nicht jedem auftreten und der Tumor selbst langsam wächst.
Wenn Symptome auftreten, hängen ihre Natur und Intensität von der Größe und dem Ort des Meningioms ab. Zusätzlich zu Kopfschmerzen und Schwindel können sie sich als epileptiforme Krämpfe, Sehbehinderung (verschwommenes Sehen), Schwäche in den Extremitäten, sensorische Störungen (Taubheit), Verlust des Gleichgewichts manifestieren.
Wenn sich ein links parietales Meningiom entwickelt, erleben Patienten: Vergesslichkeit, unstetiger Gang, Schwierigkeiten beim Schlucken, rechtsseitige motorische Schwäche mit einseitiger Muskelparalyse (Hemiparese) und Probleme mit dem Lesen (Alexia).
Ein rechtes parietales Meningiom, das zwischen den rechten parietalen Lappen und der weichen Dura (unter der Dura) bilden kann, zeigt zunächst Kopfschmerzen und bilaterale Schwäche in den Extremitäten. Eine Schwellung in der Nähe des Tumors und/oder die Komprimierung der Tumormasse in der parietalen Region kann doppelte Sehvermögen oder verschwommenes Sehen, Tinnitus und Hörverlust, Verlust des Geruchs, Anfälle sowie Sprach- und Gedächtnisprobleme verursachen. Mit einer erhöhten Kompression entwickeln einige symptome von Parietallappenläsionen auch, einschließlich parietaler Assoziationskortexdefizite mit Aufmerksamkeit oder Wahrnehmungsdefiziten; astereognose und Probleme mit der Orientierung; und kontralaterale Apraxie - Schwierigkeiten bei der Durchführung komplexer motorischer Aufgaben.
Auf der Oberfläche des Gehirns wächst ein konvexes oder konvexitisches parietales Meningiom, und mehr als 85% der Fälle sind gutartig. Zu den Anzeichen eines solchen Tumors gehören Kopfschmerzen, Übelkeit und Erbrechen, motorische Verlangsamung und häufige Muskelanfälle in Form von teilweisen Anfällen. Es kann Erosion oder Hyperostose (Verdickung des Schädels) im Schädelknochen im Kontakt mit dem Meningiom geben, und es gibt häufig eine Verkalkungszone an der Basis des Tumors, das als verkalktes Meningiom des Parietallappen definiert wird.
Komplikationen und Konsequenzen
Die Tumorvergrößerung und sein Druck können zu einem erhöhten intrakraniellen Druck, einer Schädigung der Hirnnerven (mit der Entwicklung verschiedener neurologischer Störungen), der Verschiebung und Komprimierung des parietalen Gyrus (die psychiatrische Anomalien verursachen können) führen.
Die Metastasierung ist eine äußerst seltene Komplikation, die bei Meningiomen des Grades III beobachtet wird.
Diagnose Parietale Meningeome
Die Diagnose dieser Tumoren beinhaltet eine ausführliche Anamnese der Patienten und eine neurologische Untersuchung.
Es sind Blut- und Cerebrospinalflüssigkeitstests erforderlich.
Die Hauptaufgabe bei der Tumorerkennung wird von Instrumentaldiagnostik gespielt: Gehirn CT mit Kontrast, Gehirn-MRT, MP-Spektroskopie, Positronenemissionstomographie (PET-Scan), CT-Angiographie von Hirngefäßen. [2]
Differenzialdiagnose
Die Differentialdiagnose umfasst Meningothelhyperplasie, zerebrale Tuberkulose, Gliom, Schwannom, Hemangiopericytom und alle intrakranialen mesenchymalen Tumoren.
Wen kann ich kontaktieren?
Behandlung Parietale Meningeome
Für das parietale Meningiom zielt die Behandlung darauf ab, die Kompression des Gehirns zu verringern und den Tumor zu entfernen.
Wenn der Tumor jedoch keine Symptome verursacht, erfordert er keine sofortige Behandlung: Spezialisten überwachen sein "Verhalten" mit periodischen MRT-Scans.
Für intrakranielle Meningiome chemotherapie wird selten verwendet, wenn der Tumor Grad III ist oder wiederholt ist. In den gleichen Fällen wird strahlentherapie mit einer stereotaktischen Radiochirurgie und intensitätsmodulierten Protonentherapie durchgeführt.
Die medikamentöse Behandlung, dh die Verwendung von Medizin, kann Folgendes umfassen: Einnahme eines antineoplastischen Mittel in Kapseln wie Hydroxyharnstoff (Hydroxycarbamid); Injektionen eines Antitumorhormons sandostatin. Es kann eine Immuntherapie durch Verabreichung von Alpha Interferon (2B oder 2A) Vorbereitungen angegeben werden.
Medikamente werden auch verschrieben, um einige Symptome zu lindern: Kortikosteroide zur Schwellung, Antikonvulsiva bei Anfällen usw.
Wenn ein Meningiom Symptome verursacht oder an Größe wächst, wird häufig eine chirurgische Behandlung - subtotale Resektion des Tumors - empfohlen. Während der Operation werden Proben von Tumorzellen (Biopsie) zur histologischen Untersuchung entnommen - um die Art und das Ausmaß des Tumors zu bestätigen. Obwohl eine vollständige Entfernung ein Heilmittel für das Meningiom liefern kann, ist dies nicht immer möglich. Die Position des Tumors bestimmt, wie sicher es sein wird, ihn zu entfernen. Und wenn ein Teil des Tumors bleibt, wird er mit Strahlung behandelt.
Meningiome treten manchmal nach der Operation oder Bestrahlung auf, so dass regelmäßig (alle ein bis zwei) MRIS- oder CT-Scans des Gehirns ein wichtiger Bestandteil der Behandlung sind.
Verhütung
Es gibt keine Möglichkeit, Meningiombildung zu verhindern.
Prognose
Der zuverlässigste prognostische Faktor für das parietale Meningiom ist sein histologischer Grad und das Vorhandensein von Rezidiven.
Während die 10-jährige Gesamtüberlebensrate für Grad-I-Meningiome auf fast 84% geschätzt wird, beträgt sie für Tumoren des Grades 53% (mit tödlichem Ergebnis bei Meningiomen der Grad-III). Und die Rezidivrate innerhalb von fünf Jahren nach angemessener Behandlung bei Patienten mit gutartigen Meningiomen beträgt durchschnittlich 15%, wobei atypische Tumoren - 53%und mit anaplastischen - 75% - 75%.