Facharzt des Artikels

Neue Veröffentlichungen
Apera-Syndrom
Zuletzt überprüft: 23.04.2024

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.

 [
[Ursachen aper-Syndrom
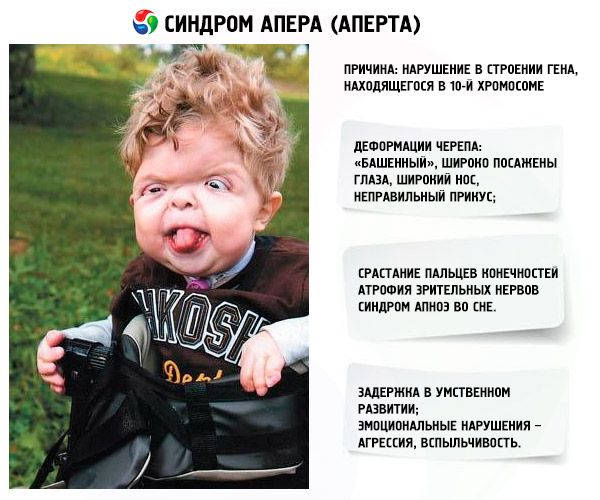
Die Entwicklung des Syndroms wird durch eine Verletzung der Struktur des im 10. Chromosom lokalisierten Gens hervorgerufen. Dieses Gen ist verantwortlich für den Prozess der Trennung der Finger und zusätzlich für den rechtzeitigen Verschluss der Schädelnähte.
Daneben das Aussehen der Pathologie verursacht bewegt als in die Tragzeit von Infektionskrankheiten (wie Tuberkulose oder Röteln, Syphilis oder Grippe und Meningitis), Mutter Röntgenbestrahlung. Meistens wird ein ähnliches Syndrom bei Kindern gefunden, die von älteren Eltern geboren wurden.
Pathogenese
Syndrom Apera wird über erblichen Weg übertragen. Sein Typ ist autosomal-dominant (dh in einer Familie, in der einer der Eltern krank ist, ist die Wahrscheinlichkeit eines Babys mit dieser Krankheit 50-100%).
Der einzigartige Wachstumsfaktor von Fibroblastenrezeptor 2 (FGFR2) aufgrund von Mutation führt zu einer Zunahme der Anzahl von Vorläuferzellen, die sich entlang des osteogenen Pfades entwickeln. Letztlich führt dies zu einer vermehrten Bildung der subperiostalen Knochenmatrix und einer vorzeitigen Ossifikation der Schädelnähte während der fetalen Entwicklung. Die Reihenfolge und Geschwindigkeit des Nahtschmelzens bestimmt den Grad der Verformung und Behinderung. Nachdem das Nahtmaterial gewachsen ist, wird das Wachstum anderer Gewebe senkrecht zu dieser Naht begrenzt und die zusammengeführten Knochen wirken als ein einzelnes Knochenskelett.
Die ersten genetischen Beweise, die bei Apert Syndrom Syndaktylien ist ein Ergebnis des Keratinozyten-Wachstumsfaktor-Rezeptor-Defekt (KGFR) wurden die Beobachtung einer Korrelation zwischen der Expression von Fibroblasten in KGFR und Schweren syndactyly.
Amblyopie und Strabismus ist häufiger bei Patienten mit der Mutation FGFR2 Ser252Trp und Atrophie der Papille ist häufiger bei Patienten mit der FGFR2 Pro253Arg-Mutation. Bei Patienten mit FGR2 Ser252Trp weisen Mutationen im Vergleich zu Patienten mit FGFR2 Pro253Arg-Mutation eine signifikant höhere Prävalenz von Sehstörungen auf.
Symptome aper-Syndrom
Einzelne Manifestationen der Krankheit sind bereits bei der Geburt des Kindes deutlich sichtbar, da sie sich sogar im Mutterleib entwickeln. Zu den wichtigsten Symptomen des Syndroms:
- Der Schädel ist deformiert - auf die Höhe gestreckt, wird zu einer Art "Turm"; Darüber hinaus sind die Augen breit gepflanzt und leicht hervorstehend (weil die Größe der Augenbahnen abnimmt); die Nase erweitert sich und ein falscher Biss entsteht (die oberen Zähne stehen übermäßig hervor);
- Die Zehen der Gliedmaßen sind vollständig verschmolzen (meist unmarkiert und auch mittig mit dem Index) und sehen aus wie eine Hautmembran oder vollständige Knochenfusion; Zusätzlich können zusätzliche Finger wachsen;
- Verzögerung der geistigen Entwicklung (überhaupt nicht);
- Die Sehnerven sind verkümmert, wodurch die Sehschärfe abnimmt (in manchen Fällen entwickelt sich ein völliger Sehverlust);
- Erhöhter intrakranieller Druck infolge zu früher Überwucherung von Schädelnähten - manifestiert sich als Kopfschmerzen und Erbrechen mit Übelkeit;
- Da der Oberkiefer unterentwickelt ist, gibt es Probleme mit dem Atmen;
- Schlafapnoe-Syndrom ist häufig.
- Emotionale Manifestationen - Aggression, Mangel an Zurückhaltung, starkes Temperament.
Komplikationen und Konsequenzen
Diagnose aper-Syndrom
Folgende Maßnahmen sind für die Diagnose erforderlich:
- Der Arzt sollte die Beschwerden des Patienten sowie die Anamnese analysieren. Es ist notwendig herauszufinden, ob es Fälle von Entwicklung einer solchen Pathologie in der Familie gegeben hat;
- Neurologische Untersuchung zur Beurteilung der Schädelform und der intellektuellen Entwicklung des Patienten (spezielle Fragebögen sowie Konversation);
- Untersuchung des Fundus, um das Vorhandensein von Symptomen eines erhöhten intrakraniellen Drucks (Ödem DZN, sowie Unschärfe seiner Ränder) zu identifizieren;
- Um den Zustand des Schädels zu beurteilen, wird seine Radiographie durchgeführt;
- Der Computer sowie Magnetresonanz-Bildgebungskopf LayerWise die zerebrale Struktur des Schädels zu untersuchen, um das Vorhandensein von Symptomen von vorzeitigem Schmelzen des Schädelnähte, um zu bestimmen, aber zusätzlich Hydrocephalus (aufgrund des erhöhten intrakraniellen Drucks ansammelt Überschuss Lauge (dieses Cerebrospinalflüssigkeit, die den Austauschprozess fördert Substanzen sowie die Ernährung des Gehirns));
- Röntgen der Füße mit Bürsten, um die Ursache herauszufinden, wegen der es eine Verschmelzung der Finger gab (dies ist wichtig für die Planung eines nachfolgenden chirurgischen Eingriffs);
- Eine Konsultation mit einem Medizingenetiker und einem Neurochirurgen kann vorgeschrieben werden.
Analysen
Die genetische Analyse der häufigen Mutationen, die im Gen des Typs FGFR2 vorkommen, wird durchgeführt.
Differenzialdiagnose
Unterscheiden Sie dieses Syndrom mit anderen genetischen Pathologien, bei denen eine Kraniosynostose beobachtet wird. Dies sind Krankheiten wie Pfeiffer, Cruson und Sethra-Chotzen und Carpenter-Syndrome. Um diese Anomalien auszuschließen, werden molekulargenetische Testmethoden verwendet.
Wen kann ich kontaktieren?
Behandlung aper-Syndrom
Die Durchführung einer Operation gilt als die einzige wirksame Methode zur Behandlung des Apera-Syndroms - sie hilft, einzelne körperliche Defekte zu korrigieren und korrigiert auch die Rückständigkeit in der geistigen Entwicklung.
Bei diesem Verfahren wird die koronale Naht geschlossen, um ein mögliches Trauma des Gehirns zu verhindern. Die häufigste Methode ist die kraniofaziale Distraktion, bei der die Abstufung des Schädels durchgeführt wird. Um einzelne Defekte im Gesicht zu entfernen, wird eine kieferorthopädische und / oder orthognathe Operation durchgeführt.
Darüber hinaus entfernen Patienten chirurgisch die Fusion der Finger.