Facharzt des Artikels

Neue Veröffentlichungen
Gemischte Bindegewebserkrankung: Ursachen, Symptome, Diagnose, Behandlung
Zuletzt überprüft: 07.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
Die Mischkollagenose ist eine seltene Erkrankung, die durch das gleichzeitige Auftreten von systemischem Lupus erythematodes, systemischer Sklerose, Polymyositis oder Dermatomyositis und rheumatoider Arthritis mit sehr hohen Titern zirkulierender antinukleärer Autoantikörper gegen Ribonukleoproteine (RNP) gekennzeichnet ist. Charakteristisch sind die Entwicklung von Handödemen, Raynaud-Syndrom, Polyarthralgie, entzündlicher Myopathie, Ösophagushypotonie und eingeschränkter Lungenfunktion. Die Diagnose basiert auf der Analyse des klinischen Bildes und dem Nachweis von Antikörpern gegen RNP in Abwesenheit von für andere Autoimmunerkrankungen charakteristischen Antikörpern. Die Behandlung ähnelt der des systemischen Lupus erythematodes und beinhaltet die Gabe von Glukokortikoiden bei mittelschwerer bis schwerer Erkrankung.
Die Mischkollagenose (MCTD) tritt weltweit bei allen Rassen auf, wobei die Häufigkeit in der Adoleszenz und im zweiten Lebensjahrzehnt am höchsten ist.
 [
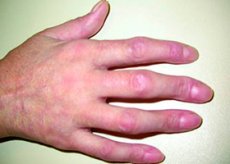
[ Klinische Manifestationen der gemischten Bindegewebserkrankung
Das Raynaud-Phänomen kann anderen Krankheitserscheinungen um mehrere Jahre vorausgehen. Oft ähneln die ersten Manifestationen einer Mischkollagenose dem Beginn eines systemischen Lupus erythematodes, einer Sklerodermie, einer rheumatoiden Arthritis, einer Polymyositis oder einer Dermatomyositis. Unabhängig von der Art der initialen Manifestationen tendiert die Krankheit jedoch dazu, fortzuschreiten und sich auszubreiten, wobei sich die Art der klinischen Manifestationen verändert.
Am häufigsten treten Ödeme an den Händen, insbesondere an den Fingern, auf, die ein wurstartiges Aussehen verursachen. Die Hautveränderungen ähneln denen bei Lupus oder Dermatomyositis. Seltener treten Läsionen auf, die denen bei Dermatomyositis ähneln, sowie ischämische Nekrose und Ulzerationen an den Fingerspitzen.
Fast alle Patienten klagen über Polyarthralgie, 75 % weisen deutliche Anzeichen einer Arthritis auf. Arthritis führt in der Regel nicht zu anatomischen Veränderungen, es können jedoch Erosionen und Deformationen auftreten, wie bei rheumatoider Arthritis. Häufig wird eine Schwäche der proximalen Muskulatur beobachtet, sowohl mit als auch ohne Schmerzen.
Nierenschäden treten bei etwa 10 % der Patienten auf und verlaufen oft mild, können aber in einigen Fällen zu Komplikationen und zum Tod führen. Bei gemischten Bindegewebserkrankungen entwickelt sich häufiger eine sensorische Neuropathie des Trigeminusnervs als bei anderen Bindegewebserkrankungen.
Diagnose einer gemischten Bindegewebserkrankung
Eine gemischte Bindegewebserkrankung sollte bei allen Patienten mit SLE, Sklerodermie, Polymyositis oder RA vermutet werden, wenn zusätzliche klinische Manifestationen auftreten. Zuallererst muss auf das Vorhandensein von antinukleären Antikörpern (ARA), Antikörpern gegen extrahierbares nukleäres Antigen und RNP getestet werden. Wenn die erhaltenen Ergebnisse mit einer möglichen MCDT übereinstimmen (z. B. sehr hoher Titer von Antikörpern gegen RNA), Gammaglobuline, Komplement, Rheumafaktor, Antikörper gegen Jo-1-Antigen (Histidyl-tRNA-Synthetase), Antikörper gegen die Ribonuklease-resistente Komponente des extrahierbaren nukleären Antigens (Sm) und DNA-Doppelhelix, sollten Antikörper getestet werden, um andere Krankheiten auszuschließen. Der Plan für weitere Untersuchungen hängt von den vorhandenen Symptomen der Organ- und Systembeteiligung ab: Myositis, Nieren- und Lungenbeteiligung erfordern die Umsetzung entsprechender Diagnosemethoden (insbesondere MRI, Elektromyographie, Muskelbiopsie).
Fast alle Patienten weisen hohe Titer (oft >1:1000) antinukleärer Antikörper auf, die mittels Fluoreszenz nachgewiesen werden. Antikörper gegen das extrahierbare nukleäre Antigen sind üblicherweise in sehr hohen Titern (>1:100.000) vorhanden. Antikörper gegen RNP sind charakteristischerweise vorhanden, während Antikörper gegen die Sm-Komponente des extrahierbaren nukleären Antigens fehlen.
Der Rheumafaktor kann in relativ hohen Titern nachgewiesen werden. Die BSG ist häufig erhöht.
Prognose und Behandlung der gemischten Bindegewebserkrankung
Die Zehnjahresüberlebensrate liegt bei 80 %, die Prognose hängt jedoch vom Schweregrad der Symptome ab. Die häufigsten Todesursachen sind pulmonale Hypertonie, Nierenversagen, Herzinfarkt, Dickdarmperforation, disseminierte Infektionen und Hirnblutungen. Bei manchen Patienten kann eine langfristige Remission auch ohne Behandlung aufrechterhalten werden.
Die Initial- und Erhaltungstherapie der Mischkollagenose ähnelt der des systemischen Lupus erythematodes. Die meisten Patienten mit mittelschwerer bis schwerer Erkrankung sprechen auf eine Glukokortikoidtherapie an, insbesondere bei frühzeitigem Beginn. Leichte Formen der Erkrankung lassen sich erfolgreich mit Salicylaten, anderen NSAR, Antimalariamitteln und in manchen Fällen mit niedrig dosierten Glukokortikoiden behandeln. Bei schwerer Organ- und Systembeteiligung sind hochdosierte Glukokortikoide (z. B. Prednisolon 1 mg/kg 1-mal täglich oral) oder Immunsuppressiva erforderlich. Entwickelt sich eine systemische Sklerose, wird eine entsprechende Therapie eingeleitet.