Facharzt des Artikels

Neue Veröffentlichungen
Hamartom
Zuletzt überprüft: 07.06.2024

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
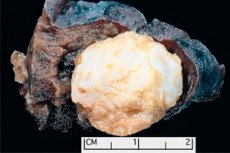
Die tumorähnliche Bildung, die in jeder anatomischen Region lokalisiert ist, die sich aus einem abnormalen Wachstum von gutartigen Gewebe ergibt, ist in der Medizin als Hamartom (aus griechischem Hamartia - Fehler, Defekt) definiert. [1]
Epidemiologie
Statistisch gesehen machen Hamartome 1,2% der gutartigen Neoplasien aus. Die Prävalenz von Lungenhamartomen wird auf ungefähr 0,25% der Allgemeinbevölkerung geschätzt und macht bis zu 8% aller Lungenneoplasmen aus. Die meisten Lungenhamartome werden bei Patienten 40 bis 70 Jahre im Alter von 40 bis 70 Jahren diagnostiziert, sind jedoch in der pädiatrischen Praxis sehr selten.
Im Allgemeinen werden die meisten Hamartome bei Männern diagnostiziert, obwohl sie in der Niere häufiger bei Frauen sind und im mittleren Alter identifiziert werden.
Etwa 5% der gutartigen Brusttumoren sind Hamartome und betreffen am häufigsten Frauen über 35 Jahre.
80-90% der Hamartomatous-Läsionen des Gehirns und mehr als 50% der Hamartome des Herzens sind mit tuberöser Sklerose verbunden.
Ursachen Hamartome
Gamartome gehören zu angeborenen Missbildungen und sind Formationen von gutartigen Charakteren, die aus mesenchymalen Geweben gebildet werden, die aus Keimeblättern stammen. Und die Ursachen ihres Auftretens sind mit einer unkontrollierten Zellteilung von zytologisch normalen Geweben (Binde, glattem Muskel, Fett oder Knorpel), charakteristisch für eine gegebene anatomische Lage und ihr fokales Überwachsen während der Embryogenese fast jeder Organ oder anatomischer Struktur verbunden.
Das Auftreten mehrerer Hamartome bei demselben Patienten wird häufig als Hamartomatose oder pleiotropes Hamartom bezeichnet.
Diese Tumoren können sporadisch oder in Gegenwart bestimmter autosomal dominanter ererbter Krankheiten sowie gentechnisch bestimmte Syndrome auftreten.
In vielen Fällen bilden sich Hamartome, wenn sich eine seltene genetische Erkrankung der multisystemischen Natur - tuberous Sklerose -kurz nach der Geburt oder bei der familiären Krankheit von Recklinghausen - neurofibromatose typ 1 manifestiert. [2]
Risikofaktoren
Zu den Hauptrisikofaktoren für die Bildung von Hamartomen gehören das Vorhandensein von sogenannten genetischen Syndromen der Hamartomatösen Polypose in der Anamnese der Patienten, einschließlich:
- Mehrfaches Hamartom-Syndrom- Cowden-Syndrom, bei dem mehrere Hamartome von ekto-, ento- und mesodermalen Ursprungs gebildet werden, werden gastrointestinale Polypose und mukokutane Manifestationen beobachtet;
- Peutz-Jeghers-Turen-Syndrom (gekennzeichnet durch die Entwicklung von gutartigen Hamartomatösen Polypen im gastrointestinalen Trakt);
- Proteus-Syndrom;
- Weil-Syndrom - juvenile Polyposis des Dickdarms;
- Das Bannayan-Riley-Ruvalcaba-Syndrom, das wie das Cowden-Syndrom mehrere Hamartome (Hamartomatöse Polypen) des Darms erzeugt;
- Carney-Stratakis-Syndrom und Carney Complex.
Darüber hinaus bilden sich Hamartome bei Patienten mit erblichem Watson-Syndrom und bei sporadisch auftretenden oder angeborenen Pallister-Hall-Syndrom mit hypothalamischem Hamartom und Polydaktylie.
Pathogenese
Der Mechanismus einer erhöhten Proliferation von Keimgeweben mit der Bildung von tumorähnlichen Missbildungen in verschiedenen Organen wird durch chromosomale Aberrationen und Genmutationen erklärt, die spontan auftreten oder vererbt werden können.
Bei tuberöser Sklerose wurden Mutationen in den TSC1- oder TSC2-Genen - Tumorsuppressoren, die eine übermäßige Proliferation verhindern und hemmen - zu schnell oder unkontrolliertes Zellwachstum und-steilung - identifiziert. Und in Neurofibromatose Typ 1 und Watson-Syndrom - Keimbahnmutationen des mitochondrialen Tumorsuppressor-Gens NF1.
Beim Hamartom-Tumorsyndrom, das Cowden, Protea, Bannayan-Riley-RUVALCABA und Juvenil-Polyposis-Syndrome kombiniert, ist die Pathogenese mit der Mutation des PTEN-Gens assoziiert, das ein Enzym codiert, das an der Regulation der Proliferation beteiligt ist, und wird als Tumor-Suppressor-Gen angesehen.
Mutationen im STK11-Gen, die die Struktur und Funktion eines der Transmembran-Serinenzyme kodieren, das seine Fähigkeit zur Einschränkung der Zellteilung zu einem Peutz-Jeghers-Turen-Syndrom verringert, mit der Entwicklung von Darmpolypen und pigmentierten Hautläsionen. Eine Mutation im GLI3-Gen, ein Transkriptionsfaktor, der an der Bildung von intrauterinem Gewebe beteiligt ist, wurde beim Pallister-Hall-Syndrom identifiziert.
Das unkontrollierte Zellwachstum aufgrund von Genmutationen führt daher zu einer Bildung von Hamartoma.
Symptome Hamartome
Abhängig von der Lokalisierung von Hamartomen sind ihre Typen unterschieden, und jeder von ihnen hat seine eigene Struktur und Symptomatik.
Hamartoma der Lunge
Lungenhamartom kann sich in jedem Lappen und peripheren Teilen der Lunge bilden und besteht aus normalen Geweben, die in der Lunge vorhanden sind: Fett, Epithel, Faser und knorpel. In 80% der Fälle vorherrscht die Chondroidkomponente (hyaline Knorpelzellen) mit der Einbeziehung von Adipozyten - Fettgewebezellen und Atemwegsepithelzellen. [3]
Die früheren Namen: Chondroid Hamartoma, Mesenchymom, chondromatöse Hamartoma oder Hamartochondrom werden derzeit nicht von WHO empfohlen.
Das mesenchymale zystische Hamartom der Lunge ist dagegen seltener und bei den meisten Patienten mit dem Cowden-Syndrom verbunden.
Die Hamartomatöse Läsion der Lunge manifestiert sich möglicherweise nicht, kann jedoch Symptome in Form von chronischem Husten (oft mit Hämoptyse) verursachen, das bei der Atmung und beim Atmen zu keuchten. [4]
Ein Hamartom des Herzens
Gutsignal Primary herztumoren zu den Erwachsenen gehören ein reifes Myozyten-Hamartom sowie bei Säuglingen und Kindern mit tuberöser Sklerose, Rhabdomyom, dh Myokardhamartom der Ventrikel oder des interventrikulären Septums. [5]
Das reife Kardiomyozyten-Hamartom entwickelt sich in der ventrikulären Wand (und selten in den Vorhöfen) und kann als mehrere Läsionen erscheinen, die dichte Massen sind, die eng mit dem zugrunde liegenden Myokard assoziiert sind. Der Tumor kann Symptome einer Herzinsuffizienz verursachen: Brustschmerzen, Herzklopfen und Arrhythmien, Herzgeräusche, Ödeme, Dyspnoe, Cyanose.
Herzhabdomyome, von denen die meisten innerhalb des ersten Lebensjahres diagnostiziert werden, bestehen aus Herzmuskelgewebe, die durch embryonale Myoblasten gebildet werden, und erscheint das Aussehen solider fokaler Massen ohne Kapsel.
Typischerweise sind diese Hamartome asymptomatisch und spontan vor dem 4-jährigen Lebensjahr zurück.
Hamartomatöse Läsionen werden auch von einigen Experten als mit dem Carney Complex myxoma of the Heart in Verbindung gebracht. [6]
Gamartoma des Magen-Darm-Trakts
Das Magenhamartom ist eine mesenchymale Masse in Form eines epithelialen hyperplastischen Polyps des Magens, Peutz-Jeghers Polyps und ein seltenes myoepitheliales Hamartom-mit hypertrophen glatten Muskelbündeln. Weitere Namen für dieses Hamartom sind Myoglandular Hamartoma, adenomyomatisches Hamartom und Magenadenomyom. Typische klinische Manifestationen umfassen Dyspepsie, epigastrische Schmerzen und Blutungen der oberen GI. [7], [8]
Weitere Informationen im Material - magenpolyposis
Ein Darmhamartom ist ein Hamartomatous oder hyperplastisches polyp des Dickdarms, der als adenomatöse oder tubuläres Adenom diagnostiziert wurde. Wenn das Hamartom in der Brunner-Drüse des Zwölffingerdrüsens lokalisiert ist, zeigen sich die Symptome durch Schmerzen in der Epigastrion. Übelkeit, Erbrechen und Blähungen (was auf Darmobstruktion hinweist); und wenn von beträchtlicher Größe, Magen-Darm-Blutungen. Bei Myoepithel-Hamartom des Ileums klagen die Patienten über Bauchschmerzen, haben ein verringertes Körpergewicht und entwickeln chronische Anämie. [9], [10]
Lesen Sie auch - rektale Polypen
Retrorektales Hamartom ist ein zystisches Hamartom oder ein mehrliebener Zyste des retorektalen Raums (das lose Bindegewebe zwischen dem Rektum und seiner eigenen Faszie), das bei Frauen mittleren Alters am häufigsten auftritt. Es ist das Aussehen einer Zyste, die aus der hinteren Wand des Rektums herausgefallen ist und mit Epithel ausgekleidet ist und chaotisch angeordnete glatte Muskelfasern enthält. Dieses Hamartom wird mit Schmerzen im unteren Bauch und wiederkehrenden Verstopfung aufweist. [11], [12]
Hamartome von Leber und Milz
Das multiple Gallenhamartom der Leber ist ein Hamartom der interdollischen intrahepatischen Gallengänge, die mit Fehlbildungen ihrer Entwicklung während der embryonalen Periode verbunden sind. Dieses Hamartom (einzeln oder mehrfach) besteht aus zufälligen, erweiterten Clustern von Gallengängen und fibrokollagenem Stroma. [13]
Gallenhamartome sind asymptomatisch und werden normalerweise übrigens (während der radiologischen Untersuchung oder Laparotomie) entdeckt. [14]
Ein seltenes und häufig nachgewiesenes primäres Neoplasma des gutartigen Charakters ist ein Hamartom der Milz, das aus Elementen des roten Zellstoffs der Milz besteht - in Form einer genau definierten homogenen Masse der festen Konsistenz. Diese Missbildung kann einzeln oder mehrfach sein; Beim Drücken des Milzparenchyms kann es im linken Subkostalbereich ein Gefühl von Beschwerden und Schmerzen geben. [15], [16]
Nieren-Hamartome
Das häufigste Hamartom der Niere wird als angiomyolipom der Niere diagnostiziert, da dieser gutartige Tumor aus reifen Fettgewebe mit eingebetteten glatten Muskeln und Blutgefäßen besteht. Es bildet in 40-80% der Fälle bei tuberöser Sklerose. Das Erhöhen der Größe des Hamartoms (mehr als 4-5 cm) führt zu Schmerzen und dem Auftreten von Blut im Urin. [17], [18]
Hamartoma der Brust
Die who-akzeptierten diagnostischen Definitionen des Brusthamartoms sind Begriffe wie Adenolipom, Chondrolipom und Myoid-Hamartom. Obwohl sie oft als Fibroadenolipom von Muammologen bezeichnet werden, weil die Tumorbildung Zellen aus faserigen, drüsen- und Fettgewebe enthält, die in einer dünnen Bindegewebekapsel mit unterschiedlichen Umrissen eingeschlossen sind. Fokale Verkalkungen können bei der Visualisierung beobachtet werden. In diesem Fall fehlen klinische Manifestationen. [19], [20]
Lesen Sie auch - brusttumoren
Hamartome des Gehirns
Ein Drittel der Patienten mit tuberöser Sklerose hat ein Hirnhamartom in Form von intrakraniellen kortikalen Auswachsen oder Tuberkeln in verschiedenen Lappen - an der Grenze der grauen und weißen Substanz - oder subependymalen Knötchen entlang der Wände der Gehirnventrikel. Astrozytisches Hamartom, ein subependymales Riesenzell-Astrozytom mit kortikaler Störung, dysmorphen Neuronen und großen Gliazellen des Gehirnparenchyms (Astrozyten), können sich ebenfalls bilden. Zu den Symptomen von Gehirnhamartomen zählen Anfälle und geistige Behinderung bei Kindern. [21], [22]
Eine seltene Fehlbildung, die während der Embryogenese auftritt und bei der Geburt vorhanden ist, ist ein hypothalamisches Hamartom, das eine Masse heterotopischer Neuronen und Gliazellen ist. Wenn das Gehirn des Kindes wächst, verteilt sich der Tumor, breitet sich jedoch nicht auf andere Gehirnregionen aus. [23], [24]
Wenn im vorderen Teil des Hypothalamus (Tuber Cinereum) hypertrophierte Gewebe gebildet werden, in der die Hypophyse daran verbindet, manifestiert Fehlbildung Symptome von zentralem frühgeborene sexuelle Entwicklung (vor 8-9 Jahren): Erscheinung von Acne-Ausschlag, frühe Entwicklung, frühe Entwicklung von Mammary und Männern und Männern, die Mammatmänner in der Male in der Male in der Male in der Male in der Male in der Male in der Male in der Male in der Male in der Male in den Frühe Schamhaar- und Sprachmutation bei Jungen.
Wenn sich Hamartome im hinteren Teil des Hypothalamus bilden, kann es in der elektrischen Aktivität des Gehirns Anomalien geben, die sich im frühen Kindheit durch Säugeln und in einem späteren Stadium (4 bis 7 Jahre) durch Epilepsie mit fokalen Epileptika-Anfällen mit plötzlichen Lachen oder mit Anbringern und Anbringern und-störungen, und-stäbchen und-stäbchen, zu korrigieren, in den Bereichen Inbetriebheit und tony und achkogn, artonischen und achkundischen, krümlichen, krümlichen und krümlichen Erkrankungen sowie ärgerlich, artonischen und adonischen und adonischen und adonischen, artonischen und adonischen und krümmten Erkenntnis manifestieren.
Ein Hypophysen-Hamartom ist ein sporadisch vorkommendes gutartiges hypophysenadenoma.
Erwachsene mittleren Alters mit Cowden-Syndrom können eine seltene tumorähnliche Masse, ein Hamartom des Kleinhirns aufweisen, das als dysplastische Kleinhirn-Gangliozytom oder Lhermitte-Duclos-Erkrankung diagnostiziert wurde. Die Symptome können fehlen oder sich als Kopfschmerzen, Schwindel, Beeinträchtigung der Koordination von Bewegungen und Lähmung einzelner Hirnnerven manifestieren.
Lymphknoten Hamartoma
Wenn die Zellen des glatten Muskel- und Fettgewebes sowie Blutgefäße und kollagenes Stroma von Inguinal-, Retroperitoneal-, Submandibulär- und Hals-Lymphknoten übertreffen, ein Angiomyomatus-Hamartom eines lynäschen Notens oder des nodulären Angiomyomatus-Hamartoms eines landes Angiomyoma-Hamartoms, das partiell oder ein vollständiger Ersatz des nodulären Angiomymaa ist. [25], [26]
Ein Hamartom der Haut
In Gegenwart von tuberöser Sklerose oder Neurofibromatose werden verschiedene Hamartome der Haut beobachtet, meistens in Form von hypopigmentierten Flecken; Kaffee und Milchflecken; angiofibroma (auf den Wangen, Kinn, nasolabialen Falten); Shagreenflecken verschiedener Lokalisierungen (die Bindegewebe-NEVI sind); faserige Plaques auf der Stirn, auf Kopfhaut oder Nacken.
Eine seltene dermatologische Manifestation von tuberöser Sklerose (insbesondere bei Männern) ist follikulocystisch und kollagen-Hamartom, die durch reichlich Kollagenablagerung in der Dermis, konzentrische perifollikuläre Fibrose und keratingefüllte, lichtschoben subkutanische Zentren gekennzeichnet ist, die auf histopathologischen Untersuchungen zu sehen waren. [27]
Zu Hamartomen bestehend aus Melanozyten (Zellen, die das Pigment-Melanin produzieren), beziehen sich die meisten Experten auch auf verschiedene melanozytische Neoplasmen, insbesondere Congenital Melanocytic Nevi
In Bezug auf die Ätiologie sind auch Hamartome, die aus Gefäßgewebe bestehen, auch hämangiome der Haut.
Patienten mit dem Peutz-Jeghers-Thuren-Syndrom haben Hamartom in Form einer fleckigen Pigmentierung der Haut und der Schleimhautmembranen - lentinose periorificialis
Fälle von linearem papulärem ektodermalem mesodermalem Hamartom (Hamartoma moniliformis) zeigen einen linearen, fleischfarbenen papulären Ausschlag am Kopf, Hals und obere Brust.
Und ein sobozytisches Hamartom ist ein Hamartom der Talgdrüsen, lesen Sie mehr in der Veröffentlichung - talg Nevus.
Hamartom des Auges
Pigmentierte hamartomatöse Läsionen der Iris bei Neurofibromatose Typ 1 und Watson-Syndrom - in Form von Knotenclustern dendritische Melanozyten - werden als Iris-Hamartome oder Lischknoten definiert. Sie sind transparent (normalerweise nicht beeinflussen) abgerundete kuppelförmige gelbbraune Papeln, die über der Oberfläche der Iris herausragen.
Und Patienten mit juvenilem Angiofibrom des Nasopharynx und familiäre adenomatöse Polyposis entwickeln oft kombiniertes Hamartom der Netzhaut und Netzhautpigmentpithel-in Form eines schwarzen Flecks auf dem zentralen (makularen) Teil der Retina. [28]
Ein Hamartom der Nase
Das Nasenhamartom wird von Spezialisten als nasales Chondromenchymal-Hamartom oder nasal-Chondrom definiert, aufgrund der gutartigen Proliferation von Atempithel, submukosaler Drüsen und Chondenbon-Bone-Mesenchyme. Seine klinischen Manifestationen hängen von der Größe und Lokalisierung der Läsion ab und umfassen: Nasenverstopfung, Schwierigkeit bei der Atmung des Nasen und des Stillens bei Säuglingen, klarer wässriger Nasenentladung und Nasenblutungen. Ein Hamartom kann mit dem Kind wachsen und sich in den Augenbahnen ausbreiten, was zu einer vorwärts gerichteten oder rückständigen Verschiebung der Störungen von Augapfel, Strabismus oder okulomotorischen Verschiebungen führt. [29]
Ein Hamartoma in einem Kind
Alle oben genannten Hamartomatösen Läsionen verschiedener Organe und anatomischer Strukturen sind bei Kindern mit entsprechenden Syndromen vorhanden.
Neugeborene treten mit mesenchymalem Hamartom der Brustwand oder des knorpeligen Hamartoms der Rippe auf, die sich auf feste unbewegliche Massen sind, die sich aus dem fokalen Überwachsen normaler Skelettelemente mit knorpeligen, vaskulären und mesenchymalen Elementen ergeben. Dieses Hamartom kann zu Atemversagen und zur Entwicklung des Atemnotsyndroms führen. Das mesenchymale Hamartom der Leber ist das zweithäufigste gutartiger Lebertumor bei Kindern. Diese tumorähnliche Bildung (häufiger im rechten Organ lokalisiert) besteht aus Zellen von mesenchymalen Stroma, Hepatozyten und Epithelzellen der Gallengangsauskleidung. Das klinische Bild umfasst tastbare Masse in der Bauchhöhle, Anorexie und Gewichtsverlust, und bei signifikanter Größe (bis zu 10 cm und mehr) deckt der Tumor extrahepatische Gallengänge und minderwertige Vena Cava ab, was zu Gelbsucht und Ödemen der unteren Extremitäten führt.
Ein Hamartom ist ein angeborenes mesoblastisches Nephrom (das bei 1 von 200.000 Säuglingen vorkommt), das im Neugeborenen mit einer tastbaren Masse dichtem Konsistenz im rechten oberen Oberquadrant des Bauches zu Bauchblähungen führen kann. Säuglinge können auch mit einer schnellen flachen Atmung auftreten.
Zu den seltenen angeborenen Anomalien gehören ein fibröses Hamartom der Kindheit, das in den ersten zwei Lebensjahren bei Kindern auftritt und als schmerzfreie knotige Masse in den subkutanen Geweben der Achselhöhle, des Nackens, der Schulter und des Unterarms, des Rückens und der Brust, des Oberschenkels, des Fußes und des externen Genitaliens darstellt.
Eccrine Angiomatous Hamartom bei einem Kind kann bei der Geburt vorhanden sein oder sich in der frühen Kindheit manifestieren. Dieser gutartige Tumor der Hamartomatous-Natur hat normalerweise das Auftreten von bläulichen oder bräunlichen Knötchen und/oder Plaques, die sich aus der Proliferation von Eccrin-Schweißdrüsengewebe und Kapillaren in den mittleren und tiefen Schichten der Dermis ergeben. Dieses Hamartom kann lokalisierte Hyperhidrose und ein erhöhtes Haarwachstum verursachen.
Komplikationen und Konsequenzen
Es ist allgemein vereinbart, dass Hamartome selten wieder auftreten oder sich in bösartige Tumoren verwandeln. Sie zeigen oft wenig oder keine Symptome und verschwinden manchmal sogar im Laufe der Zeit. In schwereren Fällen und je nach Bildungsstelle können diese Missbildungen schwerwiegende Komplikationen und Konsequenzen haben.
Zunächst kann ein Hamartoma zu einer solchen Größe wachsen, dass es auf umgebende Gewebe und Organe drückt und ihre Funktionen stört.
Herz-Hamartom bei Kindern kann zu anhaltenden Herzrhythmus-Anomalien, Klappendefekten und beeinträchtigten intrakardialen Blutfluss mit anschließender Herzinsuffizienz führen.
Komplikationen von Hamartomatous-Polypen des GI-Trakts sind gastrointestinale Blutungen, Obstruktion und Darmintussuszeption (mit möglichem tödlichem Ergebnis). Und ein großes Nierenhamartom kann die Niere brechen.
Ein Hamartom im Gehirn kann obstruktives Hydrozephalus-Syndrom verursachen.
In Hypothalamus- und Hypophysen-Hamartomen kann die Produktion von somatotropem Hormon (Wachstumshormon) beeinträchtigt werden, was zur Entwicklung von hypophysiren Nanismus (Hypopituitarismus) bei Kindern führt. Hypothalamische Hamartome bei Kindern können auch zu einer drogenresistenten Epilepsie führen.
Komplikationen des Netzhautpigmentpithelshamartoms sind mit Netzhaut- und/oder Sehnerv-Dysfunktion, Makulaödem, Neovaskularisation des Choroids und Netzhautablösung behaftet.
Diagnose Hamartome
Ein wichtiger Teil der Diagnose von Hamartomen und verwandten Syndromen ist die Sammlung von Anamnesis, einschließlich der Familiengeschichte.
Labortests umfassen Blutuntersuchungen: Allgemein klinisch; Serumelektrolyte; Lymphozytenprofil; Kalzium-, Kalium-, Phosphat- und Harnstoffspiegel; und Leberfunktionstests. Wenn möglich, erfolgt eine Punktionsbiopsie der Masse mit Feinnadelaspiration, da die histologische Untersuchung für die Diagnose und Auswahl der Behandlungstaktik von entscheidender Bedeutung ist.
Die Instrumentaldiagnostik bietet eine Visualisierung der Hamartomatous-tumorähnlichen Bildung und Identifizierung ihrer genauen Lokalisierung, für die Röntgen-, Angiographie-, Elektroenzephalographie- (EEG), Ultraschall (Sonographie), CT (Computertomographie), PET (Posistronemissionstomographie), MRT (Magnetetic Resonance Imaging) verwendet werden.
Differenzialdiagnose
In allen abnormalen Massen ist die Differentialdiagnose sehr wichtig. Somit sind Tuberkulom und Hamartom unterschieden; Lungenhamartom und primärer Lungenkrebs, bronchogenes Karzinoid, metastatische Erkrankung. Das Hirnhamartom sollte von kraniopharyngiom und hypothalamisch-chiasmatischem Gliom unterschieden werden. Und die Differentialdiagnose von Hamartom als angeborenes mesoblastisches Nephrom umfasst Wilms-Tumor (maligneres Nephroblastom), klares Zellsarkom der Nieren- und Ossifizierung von Nierentumor bei Säuglingen.
Wen kann ich kontaktieren?
Behandlung Hamartome
Wenn das Hamartom asymptomatisch ist und versehentlich entdeckt wird, ist keine Behandlung erforderlich, aber es ist notwendig, sein "Verhalten" und den Zustand des Patienten zu überwachen. In anderen Fällen zielt die Therapie darauf ab, die Intensität der Symptome zu verringern und Komplikationen zu verhindern. Beispielsweise werden bei hypothalamem Hamartom mit Symptomen vorzeitiger Pubertät bestimmte Medikamente, die die Freisetzung bestimmter Hormone hemmen, verschrieben. Herzmedikamente werden verwendet, um Symptome einer Herzinsuffizienz bei Patienten mit Herz-Hamartomen zu behandeln.
Die chirurgische Entfernung von Hamartomen ist angezeigt, um die Diagnose und bei medizinisch nicht korrigierbaren intensiven Symptomen zu bestätigen.
Beispielsweise können Lungenhamartome durch Keilresektion und in schweren Fällen durch Entfernen eines Lungenlappens (Lobektomie) reseziert werden. Ein Brusthamartom kann auch herausgeschnitten werden, und wenn es groß ist, kann eine teilweise oder vollständige Mastektomie erforderlich sein.
Die stereotaktische Hochfrequenz-Thermoablation oder Laserablation kann verwendet werden, um Hamartomatöse Polypen zu entfernen. Es wird auch eine Radiochirurgie mit hoch fokussierten Gammastrahlen - Gamma-Messer für hypothalamische Hamartome oder astrozytische Hamartome - verwendet.
Verhütung
Die einzige Methode zur Verhinderung der Entwicklung von Hamartomen kann als genetisches Screening von den zukünftigen Eltern des Kindes betrachtet werden.
Prognose
Die allgemeine Prognose dieser angeborenen Anomalie hängt von der Lokalisierung und Größe des Neoplasmas sowie von Komorbiditäten und der allgemeinen Gesundheit des Patienten ab.