Trigonozephalie
Zuletzt überprüft: 07.06.2024

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
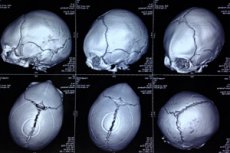
Eine angeborene Anomalie in Form einer Schädeldeformität, bei der die Köpfe von Säuglingen unregelmäßig geformt sind und der Schädel dreieckig erscheint als Trigonozephalie (aus griechischem Trigonon - Dreieck und Kephale - Kopf). [1]
Epidemiologie
Die Prävalenz von Craniosynostose wird auf etwa fünf Fälle pro 10.000 Lebendgeburten (oder einen Fall pro 2.000-2.500 in der allgemeinen Bevölkerung) geschätzt. [2]
Craniosynostose ist in 85% der Fälle sporadisch, wobei die verbleibenden Fälle als Teil eines Syndroms auftreten. [3]
Laut Statistiken ist eine vorzeitige Verschmelzung der medialen Frontalnähte die zweithäufigste Form der Craniosynostose, und die Trigonocephalie macht einen Fall pro 5.000 bis 15.000 Neugeborene aus. Die Anzahl der männlichen Säuglinge mit dieser Anomalie ist fast dreimal höher als die von weiblichen Säuglingen. [4]
In etwa 5% der Fälle ist diese angeborene Anomalie in der Familiengeschichte vorhanden. [5]
Ursachen Trigonozephalie
Die normale Bildung des Schädels erfolgt aufgrund des Vorhandenseins von Zentren des primären Wachstums und des Remodellierens von Knochen-kraniofaziale Synarthrosen (Artikulationen), die im Prozess der entwicklung des Kopfskeletts des Kopfes zu einer bestimmten Zeit zu einer bestimmten Zeit und zu einer Fusion der Knochen. [6]
Der Frontalknochen (OS Frontale) des Schädels eines Neugeborenen besteht aus zwei Hälften, zwischen denen es eine vertikale faserige Verbindung gibt-die mediale frontale oder metopische Naht (von griechischer Metopon-Stirn von griechischem Metopon). Es ist die einzige faserige Hirnnaht, die sich während der Säuglingsschaltungen übergeht: von 3 bis 4 Monaten auf 8 bis 18 Monate. [7]
Auch sehen. - schädel ändert sich nach der Geburt
Die Ursachen der Trigonozephalie sind metopisch craniosynostose (Craniostenosis) oder metopische Synostose (aus der griechischen Syntie-zusammen und Osteon-Knochen), d. H. Vor dem dritten Monat) immobile Fusion der Bones der Skull der Skull der Skull der Skull, bei der die mediale Sut der medialen Sogs miteinander miteinander miteinander in der medialen Soga fusioniert ist. Daher werden Craniosynostose und Trigonocephalie als Ursache und Wirkung oder als pathologischer Prozess und dessen Ergebnis verwandt. [8]
In den meisten Fällen ist die Trigonozephalie eines Kindes das Ergebnis einer primären (isolierten) Craniosynostose, deren genaue Ursache unbekannt ist. Isolierte Craniosynostose tritt sporadisch auf, wahrscheinlich aufgrund von Kombinationen genetischer und Umweltfaktoren. [9]
Aber die Trigonozephalie kann Teil von angeborenen Syndromen sein, die sich aus chromosomalen Anomalien und Mutationen verschiedener Gene ergeben. These include: Opitz's trigonocephaly syndrome (Boring-Opitz syndrome), aper syndrome, Loeys-Dietz syndrome, Pfeiffer syndrome, Jackson-Weiss syndrome, craniofacial dysostosis or crouzon syndrome, Jacobsen, SETRE-CHOTZEN, MUENKE-Syndrome. In solchen Fällen wird die Trigonocephalie als syndromische Trigonozephalie bezeichnet. [10]
Bei der Geburt beträgt die Größe des Gehirns normalerweise 25% seiner Erwachsenengröße und erreicht am Ende des ersten Lebensjahres etwa 75% des erwachsenen Gehirns. Bei der Verzögerung des primären Gehirnwachstums ist jedoch die sogenannte Sekundärkraniosynostose möglich. Die Ätiologie der Verzögerung ist mit Stoffwechselstörungen, einigen hämatologischen Erkrankungen und teratogenen Wirkungen auf den Fötus von Chemikalien (einschließlich der Zusammensetzung von Arzneimitteln) verbunden. [11]
Laut Experten bleibt die Trigonozephalie bei Erwachsenen, die in der Kindheit nicht durch isolierte Craniosynostose oder angeborenes Syndrom in der Kindheit behandelt wurden, das ganze Leben lang bestehen. [12]
Risikofaktoren
Spezialisten betrachten die Hauptrisikofaktoren für Trigonocephalie (und metopische Craniosynostose als Ursache) als genetisch bedingt: In den letzten zwei Jahrzehnten wurden mehr als 60 Gene identifiziert, deren Mutationen mit einer vorzeitigen unbeweglichen Fusion von Schädelknochen bei Säuglingen verbunden sind.
Es besteht ein erhöhtes Risiko für kraniofaziale Synarthrose und allgemeine Osteogenese (Knochenbildung) Abnormalitäten bei Fehlstellung des Fötus, der intrauterinen Hypoxie, mehrerer Schwangerschaften, Alkohol, Drogenkonsum oder Rauchen beim Tragen des Babys. [13]
Pathogenese
Nach der vorherrschenden Theorie liegt die Pathogenese der Trigonozephalie in einer beeinträchtigten fetalen Osteogenese in der frühen Schwangerschaft, die am häufigsten durch genetische Faktoren verursacht wird, da bei Neugeborenen mit metopischer Kraniosynostose zufällige chromosomale Anomalien nachgewiesen werden. Zum Beispiel ist Trisomie 9p eine der häufigsten und führt zu kraniofazialen und Skelettdefekten, psychomotorischen und psychomotorischen Entwicklungsverzögerungen. [14]
Aufgrund der zu frühen Verschmelzung der medialen Frontalnähte ist das Wachstum in dieser Region des Schädels schwierig: Das laterale Wachstum des Frontalknochens ist durch die Verkürzung der vorderen Schädelfossa begrenzt. Ein knöcheltes Kamm entlang der Mittellinie der Stirn bildet sich; Es gibt Konvergenz der Knochen, die die Augenbahnen und die Depression der temporalen Knochen bilden. [15]
Das Wachstum des Schädels in anderen Gebieten wird jedoch fortgesetzt: Es gibt kompensatorische Sagittal (anteroposterior) und das Querwachstum des hinteren Teils des Schädels (mit Expansion seines Parieto-Occipital-Teils) sowie des vertikalen und sagittalen Wachstums des oberen Teils des Gesichts. Infolge dieser Anomalien erwirbt der Schädel eine unregelmäßige Form - dreieckig.
Symptome Trigonozephalie
Die Hauptsymptome der Trigonozephalie sind Veränderungen in der Form und des Aussehens des Kopfes:
- Wenn der Schädel von oberhalb der Oberseite des Kopfes betrachtet wird, ist er dreieckig.
- Verengte Stirn;
- Ein prominenter oder tastbarer Grat (knöcherner Vorsprung), der entlang der Mitte der Stirn läuft, die dem frontalen Knochen eine spitze (kielige) Form verleiht;
- Verformung des oberen Teils der Augenhöhlen (Abflachung der supraorbitalen Grate) und Hypotelorismus (verringerter Abstand zwischen den Augen).
Die frontale (vordere) Fontanelle kann ebenfalls vorzeitig geschlossen sein.
In der Syndromal-Trigonocephalie gibt es andere Anomalien und Zeichen geistige Behinderung bei Kindern. [16]
Komplikationen und Konsequenzen
In schwerwiegenden Fällen wird diese angeborene Anomalie mit einem erhöhten intrakraniellen Druck begleitet, der Erbrechen, Kopfschmerzen und einen verringerten Appetit verursacht. [17]
Darüber hinaus verursacht ein erhöhter intrakranieller Druck schwere Hirnschäden, die zu kognitiven Beeinträchtigungen oder Entwicklungsverzögerung führen können. [18]
Diagnose Trigonozephalie
Die Trigonocephalie wird bei der Geburt oder innerhalb weniger Monate kurz nach der Geburt diagnostiziert. Weniger schwerwiegende Befunde der metopischen Craniosynostose können bis zur frühen Kindheit unentdeckt bleiben.
Um die Pathologie des Schädels zu visualisieren, werden instrumentelle Diagnose mit Kopf-CT-Ultraschall durchgeführt. [19], [20]
Differenzialdiagnose
Die Differentialdiagnose ist erforderlich, um einen Syndromdefekt von einer isolierten metopischen Synostose zu unterscheiden, für die dem Kind genotyp-Test verabreicht wird.
Behandlung Trigonozephalie
Bei einigen Kindern sind Fälle von metopischer Synostose ziemlich mild (wenn nur eine merkliche Furche auf der Stirn und keine anderen Symptome vorhanden ist), die keine spezifische Behandlung erfordern. [21]
Die Behandlung einer schweren Trigonozephalie ist chirurgisch und besteht aus einer Operation, um die Form des Kopfes zu korrigieren und ein normales Gehirnwachstum sowie eine chirurgische Korrektur von Gesichtsfunktionen der Gesichtsknochen zu ermöglichen. [22]
Diese chirurgische Intervention - die metopische Nahtsynostomie, die Verschiebung der Orbitalrand und die Kranioplastie - erfolgt vor dem Alter von 6 Monaten. Das Kind wird bis zum Alter von einem Jahr überwacht; In den ersten Lebensjahren wird das Kind regelmäßig untersucht, um sicherzustellen, dass es keine Sprach-, Motor- oder Verhaltensprobleme gibt. [23]
Verhütung
Die Verhinderung dieses Geburtsfehlers wurde nicht entwickelt, aber die genetische Beratung kann die Geburt eines Kindes mit einer unheilbaren kraniocerebralen Pathologie verhindern.
Und Craniosynostose im Fötus kann durch pränatale Ultraschall des Kopfes im zweiten und dritten Trimester der Schwangerschaft nachgewiesen werden.
Prognose
Die Prognose hängt weitgehend von dem Grad der Schädeldeformität ab, der die neurokognitiven Funktionen des Gehirns beeinflusst. Wenn keine Korrekturoperation durchgeführt wird, haben Kinder mit Trigonocephalie - im Vergleich zu gesunden Kollegen - insgesamt kognitive Fähigkeiten, Sprache, Sehvermögen, Aufmerksamkeit und Verhaltensprobleme.
Использованная литература