Facharzt des Artikels

Neue Veröffentlichungen
Osteoblastoklastom
Zuletzt überprüft: 07.06.2024

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
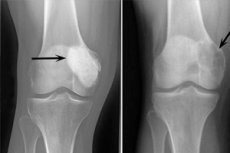
Osteoblastoklastom ist ein Tumorprozess, der entweder gutartig oder bösartig sein kann und verschiedene Skelettknochen schädigt. Zunächst wurde die Pathologie als Gigantocellular-Tumor (seit 1912) bezeichnet, 10 Jahre später schlug Dr. Stewart den Namen Osteoklastom vor. Und erst 1924 führte Professor Rusakov den raffinierten Begriff "Osteoblastoklastom" ein, der der zellulären Zusammensetzung des Neoplasmas besser entsprach.
Heute gilt Osteoblastoklastom als echtes Neoplasma, ein Weichgewebe-Tumor mit einem umfangreichen Gefäßnetzwerk. Die einzige korrekte Behandlungsoption ist die Entfernung des Tumors in gesunden Geweben, manchmal gleichzeitig mit Knochentransplantation. [1]
Epidemiologie
Die Inzidenz von Knochentumoren weltweit liegt zwischen 0,5 und 2%. Laut Statistiken der Vereinigten Staaten sind Osteosarkom (etwa 34%der Fälle), Chondrosarkom (27%) und Ewings Tumor (18-19%) am häufigsten. Chordome, Fibrosarkome, Fibrosarkome, Histiozytome, Riesenzelltumoren und Angiosarkome sind seltener.
Die Inzidenzrate korreliert stark mit dem Alter. Somit wird der erste Anstieg des Tumorwachstums im Jugendalter (etwa 16 Jahre) und der zweite Anstieg im mittleren Alter festgestellt.
Osteoblastoklastom ist ein relativ häufiger Tumor. Es tritt in etwa 2-30% aller Knochenneoplasmen auf. Frauen sind häufiger betroffen, aber Männer können auch betroffen sein, hauptsächlich im Alter zwischen 18 und 40 Jahren. Kinder unter 12 Jahren sind selten betroffen, aber selbst in diesem Alter wird die Inzidenz nicht ausgeschlossen. Es gibt Beschreibungen familiärer und erblicher Fälle von Osteoblastoklastomen.
Meistens (ca. 75%) wird der Tumor in langen Tubulusknochen gefunden, viel seltener flach und kleine Knochen sind betroffen.
In langen tubulären Knochen ist die Epimetaphyse hauptsächlich betroffen, und in der Kindheit ist die Metaphyse betroffen. Das Neoplasma sprießt nicht in den Bereich des epiphysealen und artikulären Knorpels. Sehr selten findet sich das Problem in der Diaphyse (weniger als 0,5% der Fälle).
Es wird angemerkt, dass mit der Entwicklung der Medizin die Inzidenz von Osteoblastoklastom stabil bleibt, aber die Mortalitätsraten signifikant zurückgegangen sind. Die wichtigste und wahrscheinlichste Ursache der Pathologie wird als der Einfluss ionisierender Strahlung angesehen. Daher sind die Risiken bei Menschen erhöht, die hohe Dosen an Strahlentherapie erhalten haben, sowie bei Patienten, denen Radioisotope injiziert wurden (für Diagnose oder therapeutische Zwecke). Andere gemeinsame ätiologische Faktoren sind ungünstige Ökologie und Vererbung. [2]
Ursachen Osteoblastoklastome
Osteoblastoklastom ist ein Schwerpunkt pathologisch veränderter Zellen, die in fast jedem Teil des Skeletts auftreten können. Trotz der Anomalien der Struktur teilen sich pathologische Zellen weiterhin wie in gesunden Geweben. Ihre Struktur unterscheidet sich in hohem Maße von der Norm, was den Ersatz der Eigenschaften des direkt betroffenen Knochens und seiner typischen Funktion beinhaltet. Pathologisch veränderte maligne Zellen erwerben eine Neigung zur unkontrollierten, oft schnellen Multiplikation, wobei das Tumorvolumen zunimmt. Das zuvor normale Knochengewebe kann durch die Strukturen des Neoplasmas verschoben werden, und einzelne pathologische Zellen können getrennt und mit Blut oder Lymph in andere entfernte anatomische Zonen transportiert werden. Auf diese Weise werden Metastasen gebildet.
Es ist bekannt, dass die Quelle des malignen Osteoblastoklastoms jedes maligne Neoplasma in einem beliebigen Teil des Körpers (einschließlich Tumoren innerer Organe) sein kann. Die Ausbreitung des Prozesses ist die Metastasierung. Aber die meisten Osteoblastoklastome (sowohl gutartig als auch bösartig) sind primäre Neoplasmen, die zuerst und am selben Ort auftreten und sich entwickeln.
Im Allgemeinen sind Osteoblastoklastome multifaktorielle Tumoren, deren genaue Ursachen derzeit nicht festgestellt wurden. Zu den Bedingungen für das Auftreten von Neoplasma gehören Dinge wie:
- Ein Immunschwächezustand;
- Angeborene Gewebeveränderungen;
- Mutagene Umwelteinflüsse;
- Hormonelle Veränderungen;
- Begleitende Pathologien und Verletzungen (Trauma ist häufig in der Anamnesis vorhanden).
Risikofaktoren
Es fehlen genaue Daten über die Ursachen der Osteoblastoklastombildung. Experten schlagen jedoch die Beteiligung einer Reihe von Faktoren vor, die mit einem erhöhten Risiko für Knochen-Onkopathologien verbunden sind:
- Vererbung. In vielen Fällen wird die Tendenz zu Tumorprozessen genetisch bestimmt. Insbesondere kann dies beim Leigh Fraumeni-Syndrom der Fall sein, das für die Entwicklung verschiedener Neoplasmen, einschließlich krebsartiger Tumoren und Sarkome, prädisponiert.
- Paget-Krankheit. Die Krankheit kann ein oder mehrere Knochen beeinflussen und gehört zu Pre-Tumor-Pathologien. Bei Patienten mit dieser Störung verdicken sich die Knochen und werden gleichzeitig spröde, was zu häufigen pathologischen Frakturen führt. Osteosarkome treten in etwa 8% der Fälle einer schweren Paget-Krankheit auf.
- Mehrere knöcherne Überwachsen, Exostosen.
- Mehrere Osteochondrome (einschließlich erblicher).
- Mehrere Enchondrome (das Risiko ist klein, aber immer noch vorhanden).
- Strahlenexposition (einschließlich intensiver Strahlung zur Behandlung anderer Tumorprozesse und der Auswirkungen von radioaktivem Radium und Strontium).
Eine spezielle Risikokategorie umfasst die Strahlungsbehandlung im Kindesalter und in jungen Jahren und erhält Dosen über 60 Grau.
Experten machen darauf auf die Tatsache aufmerksam, dass nichtionisierende Strahlen - insbesondere die Mikrowellen und elektromagnetische Strahlung, die aus Stromleitungen, Mobiltelefonen und Haushaltsgeräten gebildet werden - kein Risiko von Osteoblastoclastomen tragen.
Pathogenese
Die pathogenetischen Merkmale des Aussehens und der Entwicklung von Osteoblastoklastom sind nicht vollständig verstanden, was auf die Komplexität der Pathologie zurückzuführen ist. Die Grundursache für die Tumorbildung ist ein Versagen der Zelldifferenzierung aufgrund der unsachgemäßen Funktion des Immunsystems. Dies führt zum Wachstum eines Tumors, der aus "falschen", undifferenzierten Zellen besteht, die die Eigenschaften des Neoplasmas bestimmen und strukturell unreifen Zellen ähneln. Wenn die zelluläre Struktur nahezu normal ist, aber nicht, wird es als gutartiges Osteoblastoklastom bezeichnet. Bei ausgeprägten Veränderungen in der Struktur von Zellen wird der Tumor auf maligne Prozesse zurückgeführt. Für ein solches Neoplasma ist eine Veränderung der antigenen Zellfalte, des unkontrollierten Wachstums und der Zellteilung typisch. Zusammen mit dem Verlust der Spezifität der Zellstruktur leidet auch die Funktionalität. Unter anderem unterscheidet sich das maligne Osteoblastoklastom durch den Prozess der Invasion in nahe gelegene gesunde Gewebe vom gutartigen Osteoblastoklastom. Beim gutartigen Knochen-Neoplasma gibt es keine Sprosselung in gesunde Strukturen. Es besteht keine Tendenz, ein schnelles Wachstum und die Ausbreitung im gesamten Körper auszubreiten. Es gibt keine Tendenz zu willkürlicher Selbstzerstörung und Vergiftung durch Produkte der Tumorzerlegung.
Die Knochenstruktur wird in allen Fällen unabhängig von der Gütigheit der Pathologie zerstört. Infolgedessen wird das betroffene Knochensegment zerbrechlich und spröde. Oft ist der Grund, sich an Ärzte zu wenden, eine pathologische Fraktur, die auch unter minimaler Belastung auftritt.
Es ist wichtig zu beachten: Die gutartige Prozesse ist immer ein bedingter Zustand, da es Risiken der Malignisierung besteht und der gutartige Fokus transformiert wird, bösartiges Osteoblastoklastom tritt auf.
Symptome Osteoblastoklastome
Das klinische Bild im Osteoblastoklastom hängt hauptsächlich von der Lokalisierung und dem Stadium des pathologischen Prozesses ab. In der Regel wird der Tumor durch die folgenden Merkmale gekennzeichnet:
- Das Neoplasma ist einsam;
- Betrifft hauptsächlich die Rohrknochen der unteren oder oberen Gliedmaßen;
- Ist weniger häufig in flachen Knochen;
- Es gibt einen quälenden Schmerz im betroffenen Segment;
- Das Haut- und Gefäßmuster über den pathologischen Fokus nimmt zu;
- Das erkrankte Glied ist deformiert (lokalisierter Volumenerhöhung);
- Die Arbeit der Verbindung, die dem Osteoblastoclastom oder der Extremität als Ganzes am nächsten liegt, ist gestört;
- Palpatorisch ermittelte verdichteten Fokus mit einer charakteristischen "Pergamentkrise".
Im Allgemeinen können Symptome in lokale und allgemeine Symptome unterteilt werden. Lokale Symptome werden visuell nachgewiesen - insbesondere können Sie das Vorhandensein einer Krümmung oder des Ausbaus des Knochenfragments sehen. Es wird auch auf die Veränderung der Haut über den pathologischen Fokus aufmerksam gemacht: Ein Gefäßmuster manifestiert sich deutlich, die Gewebe werden geschwollen oder abgeflacht. Der Tumor kann abtastet werden - oft ist er schmerzlos, hat aber eine charakteristische Struktur. Maligne Tumoren sind typischerweise klumpig und unregelmäßig in der Konfiguration.
Das angrenzende Gelenk kann in Bewegung begrenzt sein, anhaltend schmerzhaft. Aufgrund der Komprimierung von Gefäßen und Nervenstämmen ist die Empfindlichkeit häufig beeinträchtigt und es erscheint eine anhaltende Schwellung. Das lymphatische System reagiert auch: In der Nähe von Lymphknoten werden vergrößert.
Die allgemeine Symptomatik ist eher typisch für maligne Osteoblastoklastome und ist auf die Verringerung der Körpervergiftung zurückzuführen. Patienten können:
- Fieber, fieberhafte Bedingungen;
- Hagerkeit;
- Ständige Schwäche;
- Schläfrigkeit oder Schlaflosigkeit, Appetitstörungen;
- Nacht übermäßiges Schwitzen;
- Zusammenbruch.
Es gibt auch einen kleinen Prozentsatz an Osteoblastoklastomen, die normalerweise klein und klinisch nicht ersichtlich sind. Sie werden aus anderen Gründen zu einem zufälligen Befund während radiologischer oder bildgebender Studien.
Erste Anzeichen einer Osteoblastoklastom-Ossifikation
- Beschleunigung des Wachstums des Neoplasmas.
- Erhöhtes Schmerzsyndrom.
- Expansion des destruktiven Fokus im Durchmesser oder Umwandlung der zellulären trabekulären Form in eine lytische Form.
- Zerfall der kortikalen Schicht über einen relativ langen Bereich.
- Verlust der Klarheit der Konfigurationen des zerstörerischen Fokus.
- Zerfall der Schließplatte, die zum Blockieren des Medullärkanals verwendet wurde.
- Periostale Reaktion.
Die Osteoblastoclastom-Malignität basiert auf klinischen und radiologischen Indikatoren und wird notwendigerweise durch morphologische Diagnose von Tumorgeweben bestätigt.
Zusätzlich zur Osloplastisierung eines anfänglich gutartigen Neoplasmas gibt es auch ein primäres maligne Osteoblastoclastom. Tatsächlich ist ein solcher Tumor eine Art Sarkom der osteogenen Ätiologie.
Die Lage des malignen Osteoblastoklastoms ist der gleiche wie im gutartigen Prozess. Die Radiographie zeigt einen zerstörerischen Fokus im Knochengewebe ohne klare Konturen. Die Zerstörung der kortikalen Schicht wird erweitert und wird häufig in Weichteilstrukturen beobachtet.
Anzeichen zur Unterscheidung eines malignen Osteoblastoklastoms von der osteogenen Form des osteoklastischen Sarkoms:
- Das überwiegend ältere Alter der Patienten;
- Weniger lebendige Symptomatik;
- Eine günstigere langfristige Prognose.
Osteoblastoklastom bei Kindern
Osteoblastoklastom in der Kindheit ist selten: Es gibt nur zwei oder drei Fälle pro Million Kinder. Es ist zu beachten, dass bei allen pädiatrischen Patienten diejenigen, die älter als 10-15 Jahre alt sind, vorherrschen.
Wissenschaftler können die genaue Ursache des Osteoblastoklastoms bei Kindern nicht benennen. Vermutlich ist die Pathologie mit intensivem Wachstum des Körpers des Kindes sowie mit einem genetischen Faktor verbunden.
Es gibt auch Anzeichen für mögliche Ursachen wie eine radioaktive Exposition (insbesondere Strahlentherapie), Chemotherapie (Einnahme von Zytostatik). Viele Chemotherapie-Medikamente können das genetische Material von Knochenzellen zerstören, was zur Entwicklung der Tumorentstehung führt.
Darüber hinaus ist das Risiko eines Osteoblastoklastoms bei Kindern mit bestimmten angeborenen Erkrankungen wie bilateralem Retinoblastom oder Li-Fraumeni-Syndrom höher. Eine kausale Verbindung besteht auch mit der Paget-Krankheit.
Es ist auch bekannt, dass Ärzte bei der überwiegenden Mehrheit der Kinder (etwa 90%) keine der oben genannten Risikofaktoren feststellen können.
Es ist schwierig, den Verlauf des Osteoblastoklastoms in der Kindheit vorherzusagen, da es von den Eigenschaften eines bestimmten Tumors, seiner Lokalisierung, dem Grad der Ausbreitung zum Zeitpunkt der Diagnose, der Aktualität der Behandlung und Vollständigkeit der Entfernung des Neoplasmas abhängt.
Die Qualität der Osteoblastoklastom-Behandlung hat in den letzten 2-3 Jahrzehnten große Fortschritte erzielt. Das therapeutische Protokoll ist kombiniert und die Heilungsrate ist auf mehr als 70-80%gestiegen. Ein günstiges Ergebnis kann sagen, wenn der Tumorprozess radikal chirurgisch entfernt wird und der Effekt mit einer ausreichenden Chemotherapie konsolidiert wird. Kinder mit gutartigem Osteoblastoklastom haben die beste Chance auf Genesung.
Wenn bestimmte Zahlen von gehärteten Patienten angekündigt werden, sehen wir nur allgemeine Zahlen: Keine Statistiken können die Chancen für ein bestimmtes Kind genau vorhersagen und bestimmen. Der Begriff "Genesung" wird in erster Linie als "Abwesenheit eines Tumorprozesses im Körper" verstanden, da moderne therapeutische Ansätze in der Lage sind, langfristiges Fehlen von Rezidiven sicherzustellen. Man sollte jedoch nicht die Möglichkeit unerwünschter Nebenwirkungen und verspäteten Komplikationen vergessen. Daher sollte jede Behandlung unabhängig von ihrer Komplexität in qualitativ hochwertige Rehabilitationsmaßnahmen fließen. Darüber hinaus brauchen Kinder lange Zeit noch orthopädische Versorgung.
Formen
Die Klassifizierung von Knochengewebe-Neoplasmen ist ziemlich breit. Die Aufmerksamkeit wird hauptsächlich Variationen der Zellstruktur, morphologischen Eigenschaften des Tumorprozesses geschenkt. Somit sind Tumoren in zwei Kategorien unterteilt:
- Osteogen (gebildet auf der Grundlage von Knochenzellen);
- Nesteogene (in Knochen unter dem Einfluss anderer Zelltypen gebildet - zum Beispiel vaskuläre oder Bindegewebestrukturen).
Osteoblastoklastom des Knochens ist überwiegend ein gutartiges Neoplasma. Trotzdem hat es oft ein aggressives Wachstum, trägt zur Zerstörung und Ausdünnung von Knochengeweben bei, was chirurgische Interventionen obligatorisch macht. Gleichzeitig kann auch riesige Zellosteoblastoklastom bösartig sein.
Abhängig von klinischen und radiologischen Parametern und morphologischen Bild werden drei grundlegende Formen von Osteoblastoklastomen unterschieden:
- Die zelluläre Form findet sich hauptsächlich bei älteren Menschen, sie ist durch langsame Entwicklung gekennzeichnet. Die Diagnose zeigt eine verdickte, klumpige Schwellung, ohne die Möglichkeit einer klinischen Abgrenzung des Tumorfokus aus gesunden Knochenzonen.
- Zystische Form manifestiert sich zunächst mit Schmerzen. Palpators wird das Symptom einer "Pergamentkrise" bestimmt. Visuell wird ein knöcherner Tumor aus einer glatt konvexen, kuppelförmigen Konfiguration festgestellt.
- Die lytische Form gilt als seltene Variante der Pathologie, sie wird hauptsächlich im Jugendalter nachgewiesen. Der Tumorprozess entwickelt sich schnell genug, der Patient beginnt von Schmerzen zu stören, auch bei Palpation.
Ein riesiger Zelltumor kann sich an fast jedem Knochen des Skeletts bilden, obwohl die röhrenförmigen Knochen der Gliedmaßen, Rippen und Wirbelsäule etwas häufiger betroffen sind. Osteoblastoklastom des Unterkiefers tritt zweimal so oft wie am Oberkiefer auf. Palpator ist ein dichtes Neoplasma mit weicher Zonen festgestellt. Die häufigsten Beschwerden von Patienten: Das Vorhandensein einer Ausbuchtung, die beim Kauen von Nahrung ausblutet und Beschwerden schafft. Im Verlauf des Problems wird es durch eine beeinträchtigte Funktion des Temporomandibularverbinds ergänzt. Unter den tubulären Knochen betrifft der Tumor häufiger den Femur und die Tibia. Osteoblastoklastom des Femurs ist überwiegend bei Menschen mittleren Alters zu finden. Die Krankheit wird von einer beeinträchtigten Funktion des entsprechenden Gelenks begleitet, Lahmheit tritt auf und die Haut über dem Neoplasma ist mit einem ausgeprägten Gefäßmuster bedeckt.
Zusätzlich zur obigen Klassifizierung gibt es zentrale und periphere Formen der Pathologie, obwohl es keine morphologischen Unterschiede zwischen ihnen gibt. Das periphere Osteoblastoklastom hat eine Gingivallokalisierung, und die zentrale Form entwickelt sich im Knochen und unterscheidet sich durch das Vorhandensein mehrerer Blutungen darin (daher ist der zweite Name des zentralen Osteoblastoklastoms ein brauner Tumor). Das Auftreten einer braunen Farbe ist auf die Ablagerung von Erythrozyten zurückzuführen, die sich mit der Bildung von Hämosiderin auflösen.
Maligne Knochenneoplasmen durchlaufen die folgenden Phasen ihrer Entwicklung:
- Ein T1-Foci, das 3-5 cm misst, befindet sich im Knochen und ein muskulofasziales Segment.
- Die T2-Foci erstrecken sich über den Knochenverlauf nicht mehr als 10 cm, aber nicht über einen Faszialfall hinaus.
- Die T3-Foci verlassen die Grenzen eines muskulofaszialen Gehäuses und sprießen in einen nahe gelegenen.
- Die T4-Schwerpunkte sprießen von der Haut oder den neurovaskulären Stämmen.
In ähnlicher Weise werden der Grad der Lymphknotenbeteiligung und die Ausbreitung von Metastasen kategorisiert.
Komplikationen und Konsequenzen
Zu den Komplikationen des Osteoblastoklastoms gehört eine Zunahme der Aktivität des Neoplasmas, was besonders häufig vor dem Hintergrund einer langen ruhigen Periode auftritt. In einigen solchen Fällen sprechen wir über eine bösartige Degeneration des Tumorprozesses oder über das Keimen in empfindliche anatomische Strukturen in der Nähe:
- Die Ausbreitung auf den Nervenstamm provoziert das Auftreten eines neuropathischen Schmerzsyndroms aufgrund der Auswirkung auf den Nerven groß Kaliber. Solche Schmerzen werden nach der Einnahme herkömmlicher Analgetika praktisch nicht beseitigt, so dass es den Patienten buchstäblich erschöpft.
- Die Ausbreitung auf die Blutgefäße kann durch plötzliche massive Blutungen und Hämatombildung kompliziert werden.
Komplikationen werden nicht ausgeschlossen, die von einer Verletzung der Funktion in der Nähe von Artikulationen begleitet werden: Das Wachstum des Osteoblastoklastoms in einer solchen Situation blockiert die angemessene Funktionsweise des muskuloskelettalen Mechanismus, der zu einem begrenzten Bewegungsbereich und dem Erscheinen des Schmerzsyndoms führt.
Die häufigsten Komplikationen des Osteoblastoklastoms gelten als pathologische Frakturen im betroffenen Bereich. Das Problem tritt auch bei einer geringfügigen traumatischen Wirkung auf, da das Knochengewebe extrem fragil und instabil wird.
Darüber hinaus sprechen Spezialisten auch über spezifische allgemeine und lokale Nebenwirkungen, die für bösartiges Osteoblastoklastom charakteristisch sind:
- Die Bildung von entfernten und nahen Metastasen;
- Vergiftung des Körpers mit Zerfallsprodukten.
Wenn Metastasen einige Zeit nach den anfänglichen diagnostischen Maßnahmen festgestellt werden, zeigt dies die Ineffektivität der laufenden Behandlung und des Fortschreitens des Neoplasmas an.
Eine separate Komplikationslinie ist die Entstehung neuer Tumor oder allgemeiner Pathologie aufgrund der Chemotherapie oder Bestrahlung des Osteoblastoklastom-Knochenfokus.
Diagnose Osteoblastoklastome
Zu den diagnostischen Methoden zum Nachweis von Osteoblastoclastomen gehören:
- Klinisch, einschließlich externer Untersuchung und Palpation des pathologisch veränderten Bereichs;
- Röntgen (anteroposterior und laterale Radiographie, falls angegeben - gezielte und schräge Radiographie);
- Tomographie (unter Verwendung computergestützter oder Magnetresonanztomographie);
- Radioisotop;
- Morphologisch, einschließlich histologischer, histochemischer, zytologischer Analyse von Biomaterial, die während der Punktion oder Tripanobiopsie erhalten wurden;
- Labor.
Der Arzt untersucht sorgfältig die Anamnese der Krankheit, bestimmt die ersten Anzeichen, gibt den Standort und die Art des Schmerzsyndroms an, seine Merkmale, die Ergebnisse früherer Untersuchungen und Behandlungsverfahren berücksichtigt und die Dynamik des allgemeinen Zustands des Patienten bewertet. Wenn die Pathologie von langen tubulären Knochen vermutet wird, achtet der Spezialist auf das Vorhandensein von Schwellungen, die motorische Einschränkung der engeren Artikulation sowie auf das Vorhandensein neurologischer Symptome, die Muskelschwäche und die Hypotrophie. Es ist wichtig, die internen Organe sorgfältig auf eine mögliche Ausbreitung von Metastasen auf sie zu untersuchen.
Alle Patienten führen allgemeine Blut- und Urintests mit Bestimmung von Protein- und Proteinfraktionen, Phosphor und Calcium sowie Sialinsäuren durch. Es ist auch notwendig, die enzymatische Aktivität von Phosphatasen zu bestimmen, einen bestimmten Test durchzuführen und den Index des C-reaktiven Proteins zu untersuchen. Wenn es notwendig ist, das Osteoblastoklastom vom Myloma zu differenzieren, führt der Patient einen Urintest für das Vorhandensein von pathologischem Bence-Jones-Protein durch.
Die radiologische Diagnose ist für die Diagnose eines Osteoblastoklastoms von grundlegender Bedeutung. Obligatorische überprüfte Überprüfung und gezielte Röntgenstrahlung, hochwertige Tomographie, die den Standort, die Art des pathologischen Fokus, ihre Ausbreitung auf andere Gewebe und Organe ermöglicht. Dank CT ist es möglich, den Zustand des Weichtgewebes und die dünnsten Knochenstrukturen in der notwendigen Ebene zu klären, um tiefe Schwerpunkte der pathologischen Zerstörung zu identifizieren, ihre Parameter innerhalb der Knochengrenzen zu beschreiben, um den Grad der Schädigung des umgebenden Gewebes zu bestimmen.
Gleichzeitig gilt die MRT als das informativste diagnostische Verfahren, das eine Reihe von Vorteilen sowohl gegenüber der Radiographie als auch gegenüber CT hat. Mit der Methode können Sie auch die dünnsten Gewebeschichten untersuchen und ein Bild des pathologischen Chag unter Verwendung eines räumlichen dreidimensionalen Bildes bilden.
Die obligatorische instrumentelle Diagnostik wird durch morphologische Studien dargestellt. Biomaterial wird bewertet, das während der Aspiration und Tripanobiopsie oder während der Resektion von Knochensegmenten zusammen mit dem Neoplasma erhalten wird. Die Punktionsbiopsie wird unter Verwendung von speziellen Nadeln und radiologischen Kontrolle durchgeführt.
Die folgenden Röntgenzeichen gelten als typisch für Osteoblastoclastom:
- Porositätsbeschränkung;
- Homogenität der Knochenlyse in der Art der dünnen Trabekulisierung;
- Das Vorhandensein von pseudozystischen Lucenzen, die die Struktur von eigenartigen "Seifenblasen" haben.
Dieses radiologische Bild wird von der Abwesenheit einer primären oder sekundären reaktiven osteoformativen Periostose begleitet. Ausdünnung und Atrophie der kortikalen Schicht wird festgestellt.
Die maligne Art des Osteoblastoklastoms infolge eines intensiven Gefäßlouts führt zu einer Zunahme der venösen Stasis. Gefäßveränderungen haben das Auftreten eines Neoplasmas mit reichlich vorhandener Vaskularisation.
Differenzialdiagnose
Es ist manchmal sehr schwierig, Osteoblastoclastom zu identifizieren. Probleme treten während der Differentialdiagnose der Krankheit mit osteogenem Sarkom und Knochenzysten bei Patienten unterschiedlicher Alters auf. Laut Statistiken wurde in mehr als 3% der Fälle Osteoblastoclastom mit osteogenem Sarkom und in fast 14% der Fälle für die Knochenzyste verwechselt.
Die Tabelle fasst die Hauptzeichen dieser Pathologien zusammen:
Indikatoren |
Osteoblastoclastom |
Osteogenes osteoplastisches Sarkom |
Knochenzyste |
Das häufigste Zeitalter der Inzidenz |
20 bis 30 Jahre alt |
20 bis 26 Jahre alt |
Kinder unter 14 Jahren |
Standort |
Epimetaphysealregion |
Epimetaphysealregion |
Metadiafysebereich |
Knochenrekonfiguration |
Schwere asymmetrische Ausbuchtung. |
Kleine Querausdehnung |
Eine spindelförmige Ausbuchtung. |
Konfiguration des zerstörerischen Fokus |
Die Konturen sind klar |
Die Konturen sind verschwommen, es gibt keine Klarheit |
Die Konturen sind klar |
Der Zustand des Wirbelkanals |
Bedeckt von einer Schließplatte |
An der Grenze mit dem Neoplasma geöffnet |
Keine Änderung. |
Zustand der kortikalen Schicht |
Dünn, faserig, diskontinuierlich. |
Ausdünnung, ruiniert |
Dünn, flach |
Sklerosephänomene |
Atypisch |
Gegenwärtig |
Atypisch |
Periostale Reaktion |
Abwesend |
Präsent in einer Art von "Periostal Visor" |
Abwesend |
Der Zustand der Epiphyse |
Die Lamina ist dünn, wellig. |
In der Anfangsphase bleibt ein Teil der Epiphyse intakt |
Keine Änderung. |
Knochenabschnitt in der Nähe |
Keine Änderung. |
Anzeichen von Osteoporose |
Keine Änderung. |
Obligatorische Aufmerksamkeit erfordert Indikatoren wie das Alter des Patienten, die Dauer der Pathologie, den Ort des betroffenen Fokus, andere in der Tabelle angegebene anamnestische Informationen.
Die folgenden diagnostischen Fehler sind am häufigsten, wenn das Osteoblastoklastom mit solchen pathologischen Prozessen verwechselt wird:
- Aneurysmale Zyste (lokalisiert in der Diaphyse oder Metaphyse langer tubulärer Knochen);
- Monoaxiale Art von fibröser Osteodysplasie (hauptsächlich im Kindesalter, begleitet von Knochenkrümmung ohne Knochenballon);
- Hyperparathyroid-Osteodystrophie (keine klare Abgrenzung des Fokus aus dem gesunden Knochenbereich, keine klare Knochenbucht);
- Einsame Krebs-Knochenmetastasierung (gekennzeichnet durch zerstörerische Herde mit gekrümmten "gegessenen" Konturen).
Es ist wichtig zu berücksichtigen, dass ein gutartiges Osteoblastoklastom immer verändern und bösartig werden kann. Die Ursachen der Malignität wurden noch nicht genau bestimmt, aber Wissenschaftler glauben, dass Trauma und hormonelle Veränderungen (z. B. während der Schwangerschaft) dazu beitragen. Nach einigen Beobachtungen ist auch eine Malignisierung bei wiederholten Reihe von Fernstrahlungsbehandlungen aufgetreten.
Symptome der Ossifikation:
- Das Neoplasma beginnt schnell zu wachsen;
- Der Schmerz wird schlimmer;
- Die Größe des zerstörerischen Fokus nimmt zu, und die zelluläre trabekuläre Phase übergeht in die lytische Phase;
- Die kortikale Schicht bricht zusammen;
- Die Konturen des zerstörerischen Fokus werden undeutlich;
- Die Verriegelungsplatte bricht zusammen;
- Es gibt eine periostale Reaktion.
Bei der Differenzierung des primären malignen Neoplasmas (osteogenes osteoklastisches Sarkom) und des malignen Osteoblastoklastoms wird besonderer Aufmerksamkeit auf die Dauer der Pathologie geschenkt, die Bewertung des radiologischen Bildes in der Dynamik. Auf dem Röntgenbild des primären malignen Neoplasmas gibt es keinen für Osteoblastoklastom typischen Knochenvorsprung, es gibt keine Knochenbrücken, sklerosierter Bereich mit undeutlichen Konturen können nachgewiesen werden. Bei der Malignisierung gibt es jedoch häufig einen kleinen Bereich der Verschlussplatte, der früher als Barriere für das gesunde Knochensegment diente.
Wen kann ich kontaktieren?
Behandlung Osteoblastoklastome
Die einzige korrekte Behandlung für Patienten mit Osteoblastoklastom ist die Operation. Die sanfteste Intervention findet in den Anfangsstadien der Tumorprozessentwicklung statt und stellt die Exzision der betroffenen Gewebe mit einer weiteren Füllung des Hohlraums mit einem Transplantat dar. Das Transplantat wird von einem anderen gesunden Knochen des Patienten entnommen. Eine solche Intervention ist die günstigste und weniger traumatischste, in einigen Fällen jedoch auch weniger radikal. Die Entfernung des betroffenen Knochenfragments zusammen mit dem Neoplasma wird als zuverlässigere Methode angesehen, wodurch die Wahrscheinlichkeit des Tumor-Rückwachstums auf ein Minimum reduziert wird.
Wenn es sich um ein vernachlässigter Osteoblastoklastom von großer Größe handelt, insbesondere anfällig für Malignisierung oder bereits bösartig, wird häufig eine teilweise oder vollständige Amputation der Gliedmaßen berücksichtigt.
Im Allgemeinen wird die Taktik der chirurgischen Behandlung von Osteoblastoklastom in Abhängigkeit von der Lage, der Ausbreitung und der Aggressivität des pathologischen Fokus ausgewählt.
Wenn der Tumor die langen tubulären Knochen beeinflusst, wird empfohlen, auf diese Arten von chirurgischen Eingriffen zu achten:
- Kantenresektion mit Alloplastik oder Autoplastik für gutartiger, verzögerter Prozess, Herde mit einer zellulären Struktur und in der Peripherie der Epimetaphyse. Fixierung mit Metallschrauben.
- Wenn sich das zelluläre Osteoblastoklastom auf die Mitte des Knochendurchmessers ausbreitet, werden zwei Drittel des Kondylus, teilweise der Diaphyse und der Gelenkfläche reseziert. Der Defekt ist mit artikulärem Knorpel-Allotransplantat gefüllt. Es ist fest mit Krawattenschrauben und Schrauben befestigt.
- Im Fall von Epimetaphyseverfall entlang der gesamten Länge oder der pathologischen Fraktur werden Taktiken wie eine segmentale Resektion mit artikulärer Exzision und Füllung des Defekts mit Allotransplantat verwendet. Es ist mit einer zementierten Stange fixiert.
- Bei pathologischer Fraktur und Malignisierung von Osteoblastoklastom in der proximalen Femurregion wird die gesamte Hüftendoprothetik durchgeführt.
- Bei der Resektion der Enden in der Gelenkzone des Knies wird die Technik der Allopolysubstanztransplantation mit Fixierung verwendet. Die gesamte Endoprothese mit einem erweiterten Titan-Stamm wird häufig bevorzugt, um eine nachfolgende Strahlungsbehandlung zu gewährleisten.
- Wenn sich der pathologische Fokus am distalen Ende der Tibia befindet, wird eine Resektion mit knochenplastischer Knöchelarthrodese durchgeführt. Wenn der Talusknochen betroffen ist, wird er mit der Verlängerungsarthrodese ausgerichtet.
- In Halswirbelsäulenläsionen wird ein vorderer Zugang zum C1Und C2Wirbel durchgeführt. Ein anterolateraler Zugang wird bevorzugt. Auf der Th1-th2Ebene wird ein vorderer Zugang mit schrägen Sternotomie zum dritten Interkostalraum verwendet (Gefäße werden sorgfältig nach unten verschoben). Wenn der Tumor die Körper von 3-5 Brustwirbeln beeinflusst, wird ein anterolateraler Zugang mit Resektion der dritten Rippe durchgeführt. Der Schulterblatt wird nach hinten verschoben, ohne die Muskulatur abzuschneiden. Wenn das Osteoblastoclastom in der thorakolumbalen Region zwischen Th11Und L2Gefunden wird, ist die Operation der Wahl rechtsseitig Thoracofrenolumbotomie. Der Zugang zum vorderen Teil der oberen 3 Wirbel des Kreuzbeins ist schwieriger. Ein anterolateraler retroperitonealer rechtsseitiger Zugang mit sorgfältiger Entwässerung der Gefäßstämme und Harnleiter wird empfohlen.
- Wenn die Wirbelkörper schwer zerstört werden oder die Pathologie in der Brust- und Lumbosakralwirbelsäule auf die Bogenregion ausgebreitet ist, wird in diesem Fall eine translaminare Fixierung der Wirbelsäule durchgeführt, wonach die zerstörten Wirbel mit weiteren Autoplastik entfernt werden.
- Wenn eine gutartige Form von Osteoblastoklastom in der Stirn und im Ischiasknochen nachgewiesen wird, wird das pathologisch veränderte Segment innerhalb gesunder Gewebe ohne Knochentransplantation entfernt. Wenn der Boden und das Dach des Acetabulums betroffen sind, wird eine Resektion mit weiteren Knochentransplantationen durchgeführt, um den Defekt durch Fixierung mit Spongiosis-Befestigungen zu ersetzen.
- Wenn der Iliakal, der Busen oder der Ischiasknochen betroffen sind, werden Alloplastik mit einem strukturellen Allotransplantat, einer transplantierten Osteosynthese, einer plastischen Insertion auf Zementbasis und der Neupositionierung des prothetischen Kopfes in eine künstliche Höhle durchgeführt.
- Wenn das Kreuzbein und das L2 Betroffen sind, wird eine zweistufige Intervention durchgeführt, einschließlich posteriorer Zugangsresektion des pathologisch veränderten Untersakralfragments (bis zu S2), transpedikuläre Fixierung und Entfernung des Neoplasmas von der Bone-Graftrasma mit dem Knopfmaschm mit der Knalleasma mit dem Retroperit mit dem Knochen mit dem Knochen mit Knochen mit Knochen mit dem Knochen mit dem Knochen mit dem Retroperit.
In jeder spezifischen Situation stellt der Arzt die am besten geeignete Methode der chirurgischen Intervention fest, einschließlich der Berücksichtigung der Möglichkeit, die neueste Technologie anzuwenden, um die Ergebnisse der Behandlung zu verbessern und die normale Lebensqualität des Patienten zu gewährleisten.
Verhütung
Es gibt keine spezifische Vorbeugung des Osteoblastoklastoms. Erstens ist dies auf die unzureichende Untersuchung der Ursachen der Entwicklung solcher Tumoren zurückzuführen. Viele Experten betonen die Verhinderung von Traumata zum Knochensystem unter den wichtigsten Präventivpunkten. Es gibt jedoch keine Hinweise auf den direkten Einfluss von Trauma auf die Bildung von Knochenneoplasmen, und ein Trauma lenkt in vielen Fällen nur auf den vorhandenen Tumorprozess und hat für den Ursprung des pathologischen Fokus keine offensichtliche Bedeutung, aber gleichzeitig kann es zu seinem Wachstum beitragen.
Es sollte nicht vergessen werden, dass sich häufig ein Osteoblastoklastom in Knochen bildet, die zuvor ionisierende Strahlung ausgesetzt waren - zum Beispiel zum Zweck der Therapie anderer Tumorprozesse. Radioinduzierte Neoplasien treten normalerweise nicht früher als 3 Jahre nach der Strahlungsbelastung auf.
Nicht spezifische vorbeugende Maßnahmen umfassen:
- Beseitigung schlechter Gewohnheiten;
- Einen gesunden Lebensstil führen;
- Qualität und nachhaltige Ernährung;
- Mäßige regelmäßige körperliche Aktivität;
- Vorbeugung von Verletzungen, rechtzeitige Behandlung pathologischer Prozesse im Körper, Stabilisierung der Immunität.
Prognose
Pathologische Frakturen treten häufig im betroffenen Bereich des Knochengewebes auf. In diesem Fall sind gutartige Neoplasmen, vorausgesetzt, eine radikale Behandlungsmethode hat eine günstige Prognose, obwohl die Möglichkeit von Rezidiven und Malignität des pathologischen Fokus nicht ausgeschlossen ist. Ein ungünstiges Ergebnis des gutartigen Osteoblastoklastoms ist nicht ausgeschlossen, wenn der Fokus durch aktives Wachstum und ausgeprägte Knochenzerstörung gekennzeichnet ist. Ein solcher Tumor kann ein ganzes Knochensegment schnell zerstören, was die Entwicklung einer pathologischen Fraktur und eine signifikante Beeinträchtigung der Knochenfunktion mit sich bringt. Solche Patienten haben häufig Probleme mit dem chirurgischen Ersatz des Knochengewebesfehlers und Komplikationen, die mit der Nichtheilung der Fraktur verbunden sind.
Die durchschnittliche Überlebensrate von fünf Jahren für alle Varianten maligner Osteoblastoklastome bei Kindern und Erwachsenen beträgt 70%, was als recht gut angesehen werden kann. Daher können wir zu dem Schluss kommen, dass in vielen Fällen solche Neoplasmen recht erfolgreich geheilt werden. Natürlich sind auch Punkte wie die Art des Tumorprozesses, deren Stadium, der Grad der Läsion und die Malignität von großer Bedeutung.
Offensichtlich ist es bösartiges Osteoblastoklastom, das die größte Bedrohung darstellt. In dieser Situation können wir nur bei Früherkennung, zugänglicher chirurgischer Lokalisation, Empfindlichkeit des Fokus gegenüber chemopräventiven Wirkstoffen und Strahlentherapie über eine günstige Prognose sprechen.