Facharzt des Artikels

Neue Veröffentlichungen
Polymyositis und Dermatomyositis: Ursachen, Symptome, Diagnose, Behandlung
Zuletzt überprüft: 05.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
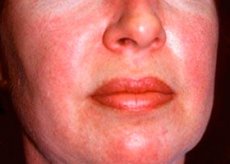
Polymyositis und Dermatomyositis sind seltene systemische rheumatische Erkrankungen, die durch entzündliche und degenerative Veränderungen der Muskulatur (Polymyositis) bzw. der Muskulatur und der Haut (Dermatomyositis) gekennzeichnet sind. Die spezifischste Hautmanifestation ist der Heliotrop-Ausschlag.
Die Muskelbeteiligung ist symmetrisch und umfasst Schwäche, leichte Druckempfindlichkeit und nachfolgende Atrophie der proximalen Beckengürtelmuskulatur. Komplikationen können viszerale Beteiligung und Malignome sein. Die Diagnose basiert auf dem klinischen Erscheinungsbild und der Beurteilung der Muskelfunktionsstörung durch Messung der Enzymwerte, MRT, Elektromyographie und Muskelbiopsie. Die Behandlung erfolgt mit Glukokortikoiden, manchmal in Kombination mit Immunsuppressiva oder intravenösen Immunglobulinen.
Frauen erkranken doppelt so häufig wie Männer. Die Krankheit kann in jedem Alter auftreten, wird jedoch am häufigsten im Alter zwischen 40 und 60 Jahren festgestellt; bei Kindern zwischen 5 und 15 Jahren.
 [
[ Was verursacht Dermatomyositis und Polymyositis?
Als Ursache der Erkrankung wird eine Autoimmunreaktion auf Muskelgewebe bei genetisch veranlagten Personen vermutet. Die Erkrankung tritt häufiger bei einer belasteten Familienanamnese und Trägern bestimmter HLA-Antigene (DR3, DR52, DR56) auf. Mögliche Auslöser sind virale Myositis und bösartige Neubildungen. Es gibt Berichte über den Nachweis von Strukturen, die Picornaviren in Muskelzellen ähneln; zudem können Viren bei Tieren ähnliche Erkrankungen auslösen. Die Assoziation bösartiger Tumoren mit Dermatomyositis (deutlich seltener als mit Polymyositis) legt nahe, dass Tumorwachstum aufgrund der Einleitung von Autoimmunreaktionen auf gemeinsame Antigene des Tumor- und Muskelgewebes ebenfalls ein Auslöser der Erkrankung sein kann.
Ablagerungen von IgM, IgG und der dritten Komplementkomponente finden sich in den Gefäßwänden der Skelettmuskulatur; dies ist besonders charakteristisch für Dermatomyositis bei Kindern. Patienten mit Polymyositis können auch andere Autoimmunprozesse entwickeln.
Pathophysiologie der Dermatomyositis und Polymyositis
Zu den pathologischen Veränderungen zählen Zellschäden und Atrophie vor dem Hintergrund unterschiedlich starker Entzündungen. Die Muskeln der oberen und unteren Extremitäten sowie des Gesichts sind weniger stark geschädigt als andere Skelettmuskeln. Schäden an den viszeralen Muskeln des Rachens und der oberen Speiseröhre, seltener an Herz, Magen oder Darm, können zu Funktionsstörungen der oben genannten Organe führen. Hohe Myoglobinkonzentrationen aufgrund von Rhabdomyolyse können Nierenschäden verursachen. Auch entzündliche Veränderungen in Gelenken und Lunge können auftreten, insbesondere bei Patienten mit Antisynthetase-Antikörpern.
Symptome von Dermatomyositis und Polymyositis
Der Beginn einer Polymyositis kann akut (insbesondere bei Kindern) oder subakut (häufiger bei Erwachsenen) sein. Eine akute Virusinfektion geht manchmal der Manifestation der Krankheit voraus oder ist deren Auslöser. Die häufigsten Manifestationen sind Schwäche der proximalen Muskulatur oder Hautausschläge. Schmerzen sind weniger ausgeprägt als Schwäche. Polyarthralgie, Raynaud-Syndrom, Dysphagie, Lungenerkrankungen und allgemeine Symptome (erhöhte Körpertemperatur, vermindertes Körpergewicht, Schwäche) können auftreten. Das Raynaud-Syndrom tritt häufig bei Patienten mit begleitenden Bindegewebserkrankungen auf.
Muskelschwäche kann sich über mehrere Wochen oder Monate verschlimmern. Damit sich Muskelschwäche klinisch manifestiert, müssen jedoch mindestens 50 % der Muskelfasern betroffen sein (das Vorhandensein von Muskelschwäche weist somit auf das Fortschreiten einer Myositis hin). Patienten können Schwierigkeiten haben, ihre Arme über Schulterhöhe zu heben, Treppen zu steigen oder aus einer sitzenden Position aufzustehen. Aufgrund einer starken Schwäche der Becken- und Schultergürtelmuskulatur können Patienten an den Rollstuhl oder das Bett gefesselt sein. Sind die Halsbeugemuskeln betroffen, wird es unmöglich, den Kopf vom Kissen zu heben. Eine Erkrankung der Rachenmuskulatur und der oberen Speiseröhre führt zu Schluckstörungen und Aufstoßen. Die Muskeln der unteren und oberen Extremitäten sowie des Gesichts sind in der Regel nicht betroffen. Es können sich jedoch Kontrakturen der Extremitäten entwickeln.
Hautausschläge bei Dermatomyositis sind meist dunkel gefärbt und erythematös. Charakteristisch ist auch ein violettes periorbitales Ödem (Heliotrop-Ekzem). Hautausschläge können leicht erhaben sein und glatt oder schuppig sein. Lokalisationen sind Stirn, Nacken, Schultern, Brust, Rücken, Unterarme, Unterschenkel, Augenbrauen, Knie, Innenknöchel, dorsale Flächen der Interphalangeal- und Metakarpophalangealgelenke sowie lateral (Gottron-Symptom). Eine Hyperämie der Nagelbasis oder -peripherie ist möglich. An der lateralen Fingerhaut kann sich eine desquamative Dermatitis mit Rissbildung entwickeln. Primäre Hautläsionen heilen oft folgenlos ab, können aber zu sekundären Veränderungen wie dunkler Pigmentierung, Atrophie, Narbenbildung oder Vitiligo führen. Subkutane Verkalkungen können sich entwickeln, insbesondere bei Kindern.
Etwa 30 % der Patienten entwickeln Polyarthralgie oder Polyarthritis, oft begleitet von Ödemen und Gelenkergüssen. Die Schwere der Gelenkmanifestationen ist jedoch gering. Sie treten häufiger auf, wenn bei Patienten Antikörper gegen Jo-1 oder andere Synthetasen nachgewiesen werden.
Eine Beteiligung innerer Organe (mit Ausnahme des Rachens und der oberen Speiseröhre) ist bei Polymyositis seltener als bei anderen rheumatischen Erkrankungen (insbesondere SLE und systemischer Sklerose). Selten, insbesondere beim Antisynthetase-Syndrom, manifestiert sich die Erkrankung als interstitielle Pneumonitis (in Form von Dyspnoe und Husten). Herzrhythmusstörungen und Reizleitungsstörungen können auftreten, verlaufen aber meist asymptomatisch. Gastrointestinale Manifestationen treten häufiger bei Kindern mit Vaskulitis auf und können zu blutigem Erbrechen, Meläna und Darmperforation führen.
Wo tut es weh?
Klassifikation der Polymyositis
Es gibt 5 Arten von Polymyositis.
- Die primäre idiopathische Polymyositis, die in jedem Alter auftreten kann, betrifft nicht die Haut.
- Die primäre idiopathische Dermatomyositis ähnelt der primären idiopathischen Polymyositis, betrifft jedoch die Haut.
- Polymyositis und Dermatomyositis im Zusammenhang mit bösartigen Neubildungen können bei Patienten jeden Alters auftreten. Ihre Entwicklung wird am häufigsten bei älteren Patienten sowie bei Patienten mit anderen Bindegewebserkrankungen beobachtet. Die Entwicklung bösartiger Neubildungen kann sowohl innerhalb von 2 Jahren vor als auch innerhalb von 2 Jahren nach Beginn der Myositis beobachtet werden.
- Eine Polymyositis oder Dermatomyositis im Kindesalter ist mit einer systemischen Vaskulitis verbunden.
- Polymyositis und Dermatomyositis können auch bei Patienten mit anderen Bindegewebserkrankungen auftreten, am häufigsten bei progressiver systemischer Sklerose, gemischter Bindegewebserkrankung und SLE.
Die Einordnung der Myositis der Rumpfmuskulatur in die Gruppe der Polymyositis ist falsch, da es sich bei letzterer um eine eigenständige Erkrankung handelt, die durch ähnliche klinische Manifestationen wie die chronische idiopathische Polymyositis gekennzeichnet ist. Sie entwickelt sich jedoch im Alter, betrifft häufig die Muskeln der distalen Körperteile (z. B. der oberen und unteren Extremitäten), hat eine längere Dauer, spricht weniger gut auf die Behandlung an und zeichnet sich durch ein typisches histologisches Bild aus.
Diagnose von Dermatomyositis und Polymyositis
Bei Patienten mit Beschwerden über proximale Muskelschwäche, mit oder ohne Druckschmerzhaftigkeit, sollte eine Polymyositis vermutet werden. Eine Abklärung auf Dermatomyositis ist bei Patienten mit einem dem Heliotrop oder Gottron-Zeichen ähnlichen Ausschlag sowie bei Patienten mit Manifestationen einer Polymyositis in Kombination mit Hautläsionen, die mit einer Dermatomyositis vereinbar sind, erforderlich. Die klinischen Manifestationen von Polymyositis und Dermatomyositis können denen einer systemischen Sklerose oder, seltener, eines systemischen Lupus erythematodes oder einer Vaskulitis ähneln. Die Diagnosesicherheit wird erhöht, wenn möglichst viele der folgenden fünf Kriterien erfüllt sind:
- Schwäche der proximalen Muskeln;
- charakteristische Hautausschläge;
- erhöhte Aktivität von Muskelgewebeenzymen (Kreatinkinase oder, falls ihre Aktivität nicht erhöht ist, Aminotransferasen oder Aldolase);
- charakteristische Veränderungen in der Myographie oder MRT;
- charakteristische histologische Veränderungen in einer Muskelgewebebiopsie (absolutes Kriterium).
Eine Muskelbiopsie kann klinisch ähnliche Erkrankungen ausschließen, wie beispielsweise eine Myositis der Rumpfmuskulatur und eine Rhabdomyolyse aufgrund einer Virusinfektion. Histologisch nachgewiesene Veränderungen können variieren, typisch sind jedoch chronische Entzündungen sowie Herde der Muskeldegeneration und -regeneration. Eine genaue Diagnose (in der Regel histologisch) ist vor Beginn einer potenziell toxischen Behandlung erforderlich. Mittels MRT können Ödeme und Entzündungen in den Muskeln nachgewiesen werden, gefolgt von einer gezielten Biopsie.
Laboruntersuchungen können den Verdacht auf das Vorliegen der Erkrankung bestätigen oder im Gegenteil ausräumen und sind auch hilfreich bei der Beurteilung ihres Schweregrads, der Möglichkeit einer Kombination mit anderen ähnlichen Pathologien und der Diagnose von Komplikationen. Obwohl bei einigen Patienten antinukleäre Antikörper nachgewiesen werden, ist dieses Phänomen typischer für andere Bindegewebserkrankungen. Etwa 60 % der Patienten haben Antikörper gegen das Antigen der Kerne (PM-1) oder ganze Thymuszellen und Jo-1. Die Rolle von Autoantikörpern in der Pathogenese der Erkrankung bleibt unklar, obwohl bekannt ist, dass Antikörper gegen Jo-1 ein spezifischer Marker des Antisynthetase-Syndroms sind, einschließlich fibrosierender Alveolitis, Lungenfibrose, Arthritis und Raynaud-Phänomen.
Die regelmäßige Bestimmung der Kreatinkinaseaktivität ist zur Therapieüberwachung hilfreich. Bei Patienten mit schwerem Muskelschwund kann die Enzymaktivität jedoch trotz chronischer aktiver Myositis normal sein. MRT, Muskelbiopsie oder erhöhte Kreatinkinaseaktivität sind oft hilfreich, um zwischen einem Rückfall der Polymyositis und einer Glukokortikoid-induzierten Myopathie zu unterscheiden.
Da bei vielen Patienten keine diagnostizierten malignen Erkrankungen vorliegen, empfehlen einige Autoren ein Screening aller Erwachsenen mit Dermatomyositis und Patienten mit Polymyositis über 60 Jahren nach folgendem Schema: körperliche Untersuchung, einschließlich Brust-, Becken- und Rektaluntersuchungen (einschließlich Stuhluntersuchung auf okkultes Blut); großes Blutbild; Blutchemie; Mammographie; karzinoembryonaler Antigentest; Urinanalyse; Röntgen-Thorax. Einige Autoren bezweifeln die Notwendigkeit eines solchen Screenings bei jüngeren Patienten ohne klinische Hinweise auf eine maligne Erkrankung.
Was muss untersucht werden?
Wie zu prüfen?
Behandlung von Dermatomyositis und Polymyositis
Bis die Entzündung abgeklungen ist, sollte die körperliche Aktivität eingeschränkt werden. Glukokortikoide sind Medikamente der ersten Wahl. Im akuten Stadium der Erkrankung sollte erwachsenen Patienten Prednisolon (oral) in einer Dosis von 40 bis 60 mg pro Tag verschrieben werden. Die regelmäßige Bestimmung der Kreatinkinase-Aktivität ist ein früher Indikator für die Wirksamkeit: Bei den meisten Patienten wird innerhalb von 6 bis 12 Wochen nach einer Zunahme der Muskelkraft eine Abnahme oder Normalisierung festgestellt. Nach Normalisierung der Enzymaktivität wird die Prednisolon-Dosis reduziert: zunächst um etwa 2,5 mg pro Tag über eine Woche, dann schneller; bei einem Rückfall der erhöhten Muskelenzymaktivität wird die Hormondosis wieder erhöht. Genesene Patienten können auf Glukokortikoide verzichten, am häufigsten benötigen erwachsene Patienten jedoch eine langfristige Glukokortikoidtherapie (10-15 mg Prednisolon pro Tag). Die Anfangsdosis von Prednisolon für Kinder beträgt 30-60 mg/m² einmal täglich. Bei Kindern kann die Glukokortikoidtherapie abgebrochen werden, wenn die Remission länger als ein Jahr anhält.
In einigen Fällen kommt es bei Patienten, die hohe Dosen Glukokortikoide erhalten, zu einer plötzlichen Zunahme der Muskelschwäche, die mit der Entwicklung einer Glukokortikoid-Myopathie verbunden sein kann.
Bei unzureichendem Ansprechen auf eine Glukokortikoid-Therapie sowie bei Auftreten einer Glukokortikoid-Myopathie oder anderer Komplikationen, die eine Dosisreduktion oder ein Absetzen von Prednisolon erfordern, sollten Immunsuppressiva (Methotrexat, Cyclophosphamid, Azathioprin, Cyclosporin) eingesetzt werden. Manche Patienten erhalten möglicherweise länger als 5 Jahre ausschließlich Methotrexat (üblicherweise in Dosen, die die zur Behandlung der rheumatoiden Arthritis verwendeten übersteigen). Intravenöse Immunglobuline können bei Patienten, die auf eine medikamentöse Therapie nicht ansprechen, wirksam sein, erhöhen aber die Behandlungskosten.
Myositiden im Zusammenhang mit primären und metastasierten Tumoren sowie Myositiden der Rumpfmuskulatur reagieren in der Regel weniger gut auf eine Glukokortikoidtherapie. Eine Remission der Myositis bei malignen Tumoren ist nach Tumorentfernung möglich.
Wie ist die Prognose bei Dermatomyositis und Polymyositis?
Bei mehr als der Hälfte der behandelten Patienten wird eine langfristige Remission (und sogar klinische Genesung) über 5 Jahre beobachtet; bei Kindern ist diese Zahl höher. Ein Rückfall kann jedoch jederzeit auftreten. Die allgemeine 5-Jahres-Überlebensrate beträgt 75 % und ist bei Kindern höher. Todesursachen bei Erwachsenen sind schwere und fortschreitende Muskelschwäche, Dysphagie, Mangelernährung, Aspirationspneumonie oder respiratorische Insuffizienz aufgrund von Lungeninfektionen. Eine Polymyositis verläuft schwerer und therapieresistenter, wenn Herz und Lunge geschädigt sind. Bei Kindern kann eine intestinale Vaskulitis zum Tod führen. Die allgemeine Prognose der Erkrankung wird auch durch das Vorhandensein bösartiger Neubildungen bestimmt.