Facharzt des Artikels

Neue Veröffentlichungen
Pierre-Robin-Syndrom
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.

Das Pierre-Robin-Syndrom, in der Medizin auch als Robin-Anomalie bekannt, ist eine angeborene Pathologie der Entwicklung des Kieferbereichs des Gesichts. Die Krankheit erhielt ihren Namen zu Ehren des französischen Zahnarztes P. Robin, der als Erster alle ihre Symptome beschrieb. Lannelongue und Menard beschrieben das Pierre-Robin-Syndrom erstmals 1891 in ihrem Bericht über zwei Patienten mit Mikrognathie, Gaumenspalte und Retroglossoptose. 1926 veröffentlichte Pierre-Robin einen Fall der Krankheit bei einem Säugling mit Symptomen des klassischen Syndroms. Bis 1974 war die Trias der Symptome als Robin-Pierre-Syndrom bekannt. Dieses Syndrom wird jedoch heute verwendet, um Fehlbildungen mit gleichzeitigem Vorhandensein mehrerer Anomalien zu beschreiben.
Epidemiologie
Es handelt sich um einen heterogenen Geburtsfehler mit einer Prävalenz von 1 zu 8.500 Lebendgeburten. Das Verhältnis von Männern zu Frauen beträgt 1:1, mit Ausnahme der X-chromosomalen Form.
Unter diesen Patienten haben 50 % der Säuglinge eine unvollständige Gaumenspalte, der Rest wird mit einem gewölbten und ungewöhnlich hohen Gaumen, aber ohne Spalte geboren.
Ursachen Pierre-Robin-Syndrom
Die Möglichkeit einer autosomal-rezessiven Vererbung der Krankheit wird in Betracht gezogen. Je nach Ätiologie gibt es zwei Arten von Syndromen: isolierte und genetisch bedingte. Der isolierte Typ entsteht durch Kompression des Unterkiefers während der Embryonalentwicklung. Kompression kann entstehen durch:
- Das Vorhandensein lokaler Versiegelungen in der Gebärmutter (Zysten, Narben, Tumore).
- Mehrlingsschwangerschaft.
Außerdem kann die Kieferentwicklung des Fötus gestört sein durch:
- Virusinfektionen, die die werdende Mutter während der Schwangerschaft erlitten hat.
- Neurotrophe Störungen.
- Unzureichende Menge an Folsäure im Körper einer schwangeren Frau.
Pathogenese
Das Pierre-Robin-Syndrom ist auf embryonale Störungen zurückzuführen, die durch eine Vielzahl von Pathologien in der pränatalen Phase verursacht werden.
Es gibt drei pathophysiologische Theorien, die das Auftreten des Pierre-Robin-Syndroms erklären könnten.
Mechanische Theorie: Diese Theorie ist die wahrscheinlichste. Eine Unterentwicklung des Kieferapparates tritt zwischen der 7. und 11. Schwangerschaftswoche auf. Die hohe Position der Zunge in der Mundhöhle führt zur Bildung von Gaumenspalten, wodurch sich die Hohlvene nicht schließt. Diese Theorie erklärt die klassische umgekehrte U-förmige Spalte und das Fehlen der zugehörigen Lippenspalte. Ein Oligohydramnion kann ätiologische Bedeutung haben, da fehlendes Fruchtwasser zu einer Deformation des Kinns und einer anschließenden Kompression der Zunge zwischen der Hohlvene führen kann.
Neurologische Theorie: Bei der Elektromyographie der Muskeln des Zäpfchens und der Rachensäule wurde eine Verzögerung der neurologischen Entwicklung festgestellt, und aufgrund einer Leitungsverzögerung im Nervus hypoglossus kam es zu Geschmacksstörungen.
Theorie der Dysneuroregulation des Rautenhirns: Diese Theorie basiert auf der Störung der Entwicklung des Rautenhirns während der Ontogenese.
Eine unzureichende Entwicklung des Unterkiefers führt zu einer deutlichen Verkleinerung der Mundhöhle. Dies wiederum führt zur sogenannten Pseudomakroglossie, d. h. die Zunge verlagert sich an die Rückseite der Rachenwand. Diese Pathologie führt zur Entwicklung einer Atemwegsobstruktion.
Solange das Baby weint oder sich bewegt, bleiben die Atemwege frei, doch sobald das Baby einschläft, kommt es erneut zu einer Obstruktion.
Aufgrund von Atemwegserkrankungen ist das Füttern des Babys sehr schwierig. Zu diesem Zeitpunkt kommt es fast immer zu einer Obstruktion der Atemwege. Ohne medizinische Behandlung kann eine solche Pathologie zu schwerer Erschöpfung des gesamten Körpers und sogar zum Tod führen.
Symptome Pierre-Robin-Syndrom
Die Krankheit ist durch drei Hauptsymptome gekennzeichnet:
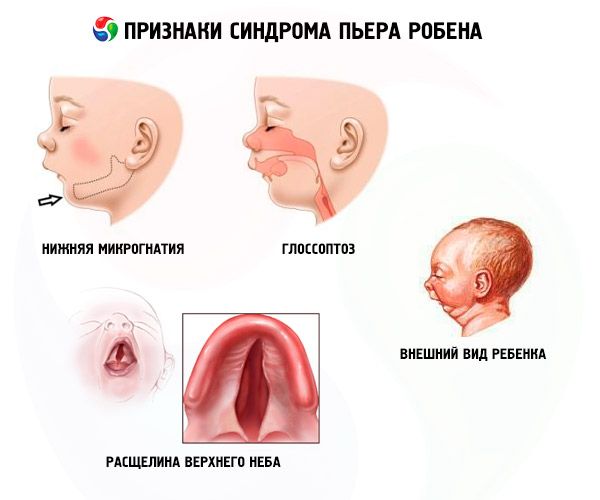
- Untere Mikrognathie (Unterentwicklung des Unterkiefers, tritt in 91,7 % der Krankheitsfälle auf). Sie ist durch eine Retraktion des unteren Zahnbogens um 10–12 mm hinter den oberen Zahnbogen gekennzeichnet. Der Unterkiefer hat einen kleinen Körper und einen stumpfen Winkel. Das Kind erreicht im Alter von etwa 5–6 Jahren eine normale Entwicklung.
- Glossoptose (Zurückziehen der Zunge aufgrund unzureichender Entwicklung, wird in 70–85 % der Fälle beobachtet).
- Makroglossie und Ankyloglossie sind relativ seltene Symptome, die in 10–15 % der Fälle auftreten.
- Ein Riss erscheint im Himmel.
- Bradypnoe und Dyspnoe.
- Leichte Zyanose.
- Asphyxie, die am häufigsten beim Versuch auftritt, das Baby zu füttern.
- Das Schlucken ist unmöglich oder sehr schwierig.
- Ich habe das Gefühl, mich übergeben zu müssen.
- Ohrmuschelanomalien in 75 % der Fälle.
- Bei 60 % der Patienten kommt es zu einer Schallleitungsschwerhörigkeit, während eine Atresie des äußeren Gehörgangs nur bei 5 % der Patienten auftritt, d. h. eine unzureichende Pneumatisierung der Warzenhöhle des Schläfenbeins.
- Anomalien des Innenohrs (Aplasie der seitlichen Bogengänge, großer Aquaeductus vestibularis, Verlust der Cochlea-Haarzellen).
- Nasenfehlbildungen sind selten und bestehen hauptsächlich aus Anomalien der Nasenwurzel.
- Zahnfehlbildungen treten in 30 % der Fälle auf. Laryngomalazie und Velopharyngealinsuffizienz treten bei etwa 10–15 % der Patienten mit Pierre-Robin-Syndrom auf.
Systemische Merkmale des Pierre-Robin-Syndroms
In 10–85 % der registrierten Fälle werden systemische Entwicklungsanomalien beschrieben.
Bei 10–30 % der Patienten treten Augenanomalien auf. Dazu gehören beispielsweise Weitsichtigkeit, Kurzsichtigkeit, Astigmatismus, Hornhautsklerose und Stenose des Tränennasengangs.
Herz-Kreislauf-Erkrankungen: gutartige Herzgeräusche, Pulmonalarterienstenose, offener Ductus arteriosus, ovales Fenster, Vorhofseptumdefekt und pulmonale Hypertonie. Ihre Prävalenz variiert zwischen 5 und 58 %.
Anomalien des Bewegungsapparates (70–80 % der Fälle): Syndaktylie, dysplastische Phalangen, Polydaktylie, Klinodaktylie, Hypermobilität der Gelenke und Oligodaktylie der oberen Extremitäten. Anomalien der unteren Extremitäten: Fußanomalien (Klumpfuß, Metatarsaladduktion), Oberschenkelfehlbildungen (Valgus- oder Varusstellung des Beckens, kurze Oberschenkelknochen), Hüftfehlbildungen (angeborene Luxation, Kontrakturen), Kniegelenkfehlbildungen (GENU VALGUS, Synchondrose). Fehlbildungen der Wirbelsäule: Skoliose, Kyphose, Lordose, Wirbeldysplasie, Agenesie des Kreuzbeins und des Steißbeinsinus.
Pathologie des zentralen Nervensystems: Epilepsie, Verzögerungen in der Entwicklung des Nervensystems, Hydrozephalus. Die Häufigkeit von ZNS-Defekten beträgt etwa 50 %.
Urogenitalanomalien: Hodenhochstand (25 %), Hydronephrose (15 %) und Hydrozele (10 %).
Assoziierte Syndrome und Erkrankungen: Stickler-Syndrom, Trisomie 11q-Syndrom, Trisomie 18, 4q-Deletionssyndrom, rheumatoide Arthropathie, Hypochondroplasie, Moebius-Syndrom.
Bühnen
Es gibt drei Schweregrade der Erkrankung, die vom Zustand der Atemwege des Kindes abhängen:
- Leicht – es gibt geringfügige Probleme beim Füttern, aber das Atmen ist fast nicht schwierig. Die Behandlung erfolgt ambulant.
- Mittelschwer – Das Atmen ist mittelschwer, das Füttern des Kindes mittelschwer. Die Behandlung erfolgt im Krankenhaus.
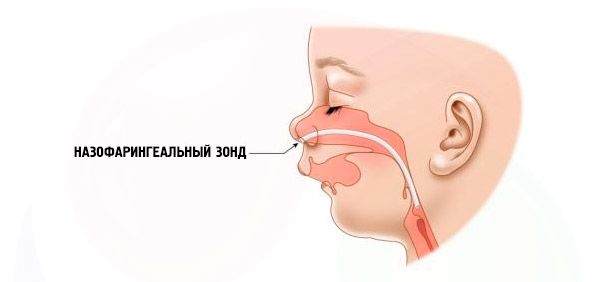
- Schwerwiegend – Das Atmen fällt sehr schwer, das Kind kann nicht normal ernährt werden. Es ist notwendig, spezielle Geräte (intranasale Sonde) zu verwenden.
Komplikationen und Konsequenzen
Die Kombination aus Mikrognathie und Glossoptose kann zu schweren Atemwegskomplikationen und Problemen beim Füttern des Kindes führen.
Das Pierre-Robin-Syndrom verursacht folgende Komplikationen:
- Stridose Atmung aufgrund einer Atemwegsobstruktion. Laryngomalazie oder sogar Schlafasphyxie.
- Die psychomotorische Entwicklung des Kindes hinkt der seiner Altersgenossen weit hinterher.
- Auch die körperliche Entwicklung bleibt zurück.
- Die Sprache der Patienten ist beeinträchtigt.
- Häufige Ohrenentzündungen, die chronisch werden und zu Hörschäden führen.
- Obstruktives Schlafapnoe-Syndrom, das Auftreten des Todes im Schlaf variiert in 14-91 % der Fälle.
- Probleme mit den Zähnen.
Diagnose Pierre-Robin-Syndrom
Die Diagnose des Pierre-Robin-Syndroms ist nicht schwierig. Sie basiert auf klinischen Manifestationen. Um andere Erkrankungen auszuschließen, ist die Konsultation eines Genetikers sehr wichtig.
Kinder mit Robins angeborener Anomalie haben von Geburt an Atemprobleme, da die Zunge ständig zurücksinkt. Das Baby ist unruhig, seine Haut ist bläulich, beim Einatmen keucht es aus der Brust. Beim Füttern kann es zum Ersticken kommen. Die Diagnose kann auch durch das ungewöhnliche Aussehen des Kindes gestellt werden – das „Vogelgesicht“. Häufig entwickeln Patienten weitere Defekte: Myopie, Katarakt, Erkrankungen des Urogenitalsystems, Herzerkrankungen, Anomalien in der Entwicklung der Wirbelsäule.
Aufgrund dieser klinischen Manifestationen wird es für einen Spezialisten nicht schwierig sein, eine korrekte Diagnose zu stellen.
Wen kann ich kontaktieren?
Behandlung Pierre-Robin-Syndrom
Die Behandlung erfolgt unmittelbar nach der Geburt eines Kindes mit Pierre-Robin-Syndrom. Bei leichter Erkrankung ist es zur Verbesserung des Zustands des Patienten notwendig, das Kind ständig aufrecht oder auf dem Bauch liegend zu halten. Der Kopf des Babys sollte zur Brust geneigt sein. Es wird nicht empfohlen, das Kind während des Fütterns horizontal zu halten, damit keine Nahrung in die Atemwege gelangt.
Bei ausgeprägter Unterentwicklung des Unterkiefers wird die zurückgezogene Zunge durch einen chirurgischen Eingriff in eine normale physiologische Position gebracht. In schweren Fällen wird die Zunge nach oben gezogen und an der Unterlippe fixiert. In sehr schweren Fällen müssen eine Tracheotomie, eine Glossopexie und eine Distraktionsosteogenese des Unterkiefers durchgeführt werden.
Auch konservative Behandlungen kommen zum Einsatz.
Medikamente
Phenobarbital. Schlaf- und Beruhigungsmittel mit krampflösender Wirkung. Jede Tablette enthält 100 ml Phenobarbital. Die Dosierung ist individuell und hängt vom Schweregrad der Erkrankung und dem Zustand des Kindes ab. Das Medikament ist verboten bei Patienten mit Leberversagen, Hyperkinese, Anämie, Myasthenie, Porphyrie, Diabetes mellitus, Depression und Unverträglichkeit der Inhaltsstoffe. Folgende Symptome sind bei der Einnahme möglich: Schwindel, Asthenie, Halluzinationen, Agranulozytose, Übelkeit, niedriger Blutdruck und Allergien.
Clonazepam. Ein Medikament zur Behandlung von Epilepsie. Das Medikament enthält den Wirkstoff Clonazepam, ein Benzodiazepin-Derivat. Es wirkt krampflösend, angstlösend und muskelentspannend. Die Dosis wird vom behandelnden Arzt festgelegt, sollte jedoch maximal 250 µg pro Tag nicht überschreiten. Nicht einnehmen bei Schlaflosigkeit, Muskelhypertonie, psychomotorischer Unruhe oder Panikstörungen. Folgende Symptome sind bei der Einnahme möglich: Lethargie, Übelkeit, Dysmenorrhoe, Kopfschmerzen, Leukopenie, Harnverhalt oder Inkontinenz, Alopezie, Allergie.
Erhältlich als Lösung und als Rektaltablette. Der Wirkstoff ist ein Benzodiazepin-Derivat (Sibazon). Es wirkt beruhigend, angstlösend und krampflösend. Die Dosierung ist individuell. Patienten mit chronischer Hyperkapnie, Myasthenie und Benzodiazepin-Intoleranz ist die Einnahme des Arzneimittels untersagt. Bei der Anwendung des Arzneimittels können folgende Symptome auftreten: Übelkeit, Verstopfung, Kopfschmerzen, Schwindel, Schluckauf, Harninkontinenz, Allergien.
Cortexin-Lyophilisat. Ein Medikament mit nootropischer Wirkung. Das Medikament enthält einen Komplex aus wasserlöslichen Polypeptidfraktionen und Glycin. Die Dosierung ist individuell und wird vom behandelnden Arzt entsprechend dem Zustand des Patienten verordnet. Patienten mit einer Cortexin-Unverträglichkeit ist die Einnahme des Medikaments untersagt. Das Medikament kann allergische Reaktionen hervorrufen.
Physiotherapeutische Behandlung
In leichten Stadien des Syndroms wird typischerweise eine Lagerungstherapie angewendet, bei der das Kind in aufrechter Position auf den Bauch gelegt wird, bis die Schwerkraft den Unterkiefer zum richtigen Wachstum zwingt.
Chirurgische Behandlung
Die Korrektur einer Glossoptose erfolgt in erster Linie durch eine chirurgische Behandlung. Es gibt verschiedene Methoden:
- Unterstützung der Zunge mit einem Silberfaden. Der Faden wird durch den unteren Teil des Zahnfleisches und die Unterlippe geführt. Die Methode heißt Douglas.
- Duhamels Methode: Ein dicker Silberfaden wird durch die Zungenwurzel und beide Wangen des Patienten geführt. Die Anwendung sollte nicht länger als 30 Tage dauern.
- Orthopädische Geräte zur Zungenverlängerung und -fixierung.
- Im Alter von einem Jahr kann eine Operation zur Korrektur einer Gaumenspalte durchgeführt werden.
Prognose
Prognose und Verlauf der Erkrankung sind schwerwiegend. Am häufigsten tritt der Tod in den ersten Lebenstagen im mittelschweren bis schweren Stadium der Erkrankung ein (Ursache ist Asphyxie). Zudem ist das Sterberisiko im ersten Lebensjahr aufgrund zahlreicher Infektionen recht hoch.
Bei Patienten über zwei Jahren ist die Prognose günstig.
 [ 36 ]
[ 36 ]