Facharzt des Artikels

Neue Veröffentlichungen
Aper-Syndrom
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.

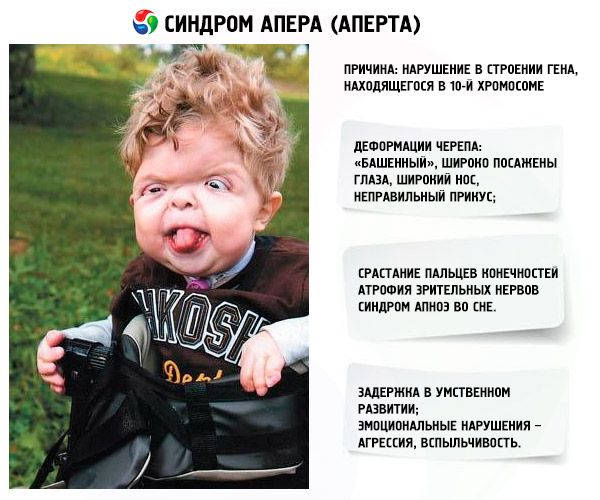
Das Apert-Syndrom ist eine genetische Erkrankung, die zu Entwicklungsdefekten des Schädels (weit auseinanderstehende Augen, übermäßig hoher Schädel) und darüber hinaus der unteren und oberen Gliedmaßen (die Finger sind vollständig verwachsen, es können auch zusätzliche Finger auftreten) führt.
 [
[ Ursachen Aper-Syndrom
Die Entstehung des Syndroms wird durch eine Störung in der Struktur eines Gens auf Chromosom 10 hervorgerufen. Dieses Gen ist für den Prozess der Fingertrennung und darüber hinaus für den rechtzeitigen Verschluss der Schädelnähte verantwortlich.
Darüber hinaus gelten Infektionskrankheiten während der Schwangerschaft (wie Röteln oder Tuberkulose, Syphilis oder Grippe sowie Meningitis) sowie die Exposition der Mutter gegenüber Röntgenstrahlen als Ursachen der Pathologie. Am häufigsten wird ein solches Syndrom bei Kindern älterer Eltern festgestellt.
Pathogenese
Das Apert-Syndrom ist erblich. Sein Typ ist autosomal-dominant (d. h. in einer Familie, in der ein zukünftiger Elternteil krank ist, beträgt die Wahrscheinlichkeit, ein Baby mit dieser Krankheit zu bekommen, 50-100 %).
Eine einzigartige Mutation im Fibroblasten-Wachstumsfaktor-Rezeptor 2 (FGFR2) führt zu einer erhöhten Anzahl von Progenitorzellen, die sich entlang des osteogenen Pfades entwickeln. Dies führt letztendlich zu einer verstärkten subperiostalen Knochenmatrixbildung und einer vorzeitigen Verknöcherung der Schädelnähte während der fetalen Entwicklung. Die Reihenfolge und Geschwindigkeit des Schmelzens der Naht bestimmen den Grad der Deformität und Behinderung. Sobald das Nahtmaterial verheilt ist, wird das Wachstum anderer Gewebe senkrecht zu dieser Naht eingeschränkt, und die verschmolzenen Knochen fungieren als einheitliches Knochengerüst.
Der erste genetische Beweis dafür, dass die Syndaktylie beim Apert-Syndrom auf einen Defekt im Keratinozyten-Wachstumsfaktor-Rezeptor (KGFR) zurückzuführen ist, war die Beobachtung einer Korrelation zwischen der KGFR-Expression in Fibroblasten und dem Schweregrad der Syndaktylie.
Amblyopie und Strabismus treten häufiger bei Patienten mit der FGFR2 Ser252Trp-Mutation auf, und eine Sehnervenatrophie tritt häufiger bei Patienten mit der FGFR2 Pro253Arg-Mutation auf. Patienten mit FGR2 Ser252Trp-Mutation weisen im Vergleich zu Patienten mit der FGFR2 Pro253Arg-Mutation eine signifikant höhere Prävalenz von Sehbehinderungen auf.
Symptome Aper-Syndrom
Einige Manifestationen der Krankheit sind bereits bei der Geburt des Babys deutlich erkennbar, da sie sich im Mutterleib entwickeln. Zu den Hauptsymptomen des Syndroms gehören:
- Der Schädel ist deformiert – er dehnt sich in die Höhe und nimmt das Aussehen eines „Turms“ an; außerdem stehen die Augen weit auseinander und treten leicht hervor (weil die Größe der Augenhöhlen abnimmt); die Nase verbreitert sich und es bildet sich ein falscher Biss (die oberen Zähne stehen übermäßig hervor);
- Die Finger der Gliedmaßen sind vollständig miteinander verwachsen (hauptsächlich der Ringfinger sowie der Mittelfinger und der Zeigefinger) und sehen aus wie eine Hautmembran oder eine vollständige Knochenfusion; außerdem können zusätzliche Finger wachsen;
- Geistige Behinderung (tritt nicht bei jedem auf);
- Die Sehnerven verkümmern, wodurch die Sehschärfe abnimmt (in einigen Fällen kommt es zum vollständigen Verlust des Sehvermögens);
- Erhöhter Hirndruck, der durch vorzeitigen Verschluss der Schädelnähte entsteht, äußert sich in Form von Kopfschmerzen und Erbrechen mit Übelkeit;
- Da der Oberkiefer unterentwickelt bleibt, kommt es zu Atemproblemen;
- Das Schlafapnoe-Syndrom kommt häufig vor.
- Emotionale Manifestationen – Aggression, mangelnde Zurückhaltung, starke Reizbarkeit.
Komplikationen und Konsequenzen
Bei den meisten Patienten kommt es im Säuglingsalter zu einer Obstruktion der oberen Atemwege. Aufgrund der geringen Größe des Nasenrachenraums und der Choanen sind die oberen Atemwege schlecht zugänglich, und die unteren Atemwege können aufgrund von Anomalien des Trachealknorpels zum frühen Tod führen.
Diagnose Aper-Syndrom
Um eine Diagnose zu stellen, sind folgende Schritte erforderlich:
- Der Arzt sollte die Beschwerden des Patienten sowie die Krankengeschichte analysieren. Es ist notwendig herauszufinden, ob es in der Familie Fälle ähnlicher Pathologie gab.
- Neurologische Untersuchung zur Beurteilung der Schädelform und der intellektuellen Entwicklung des Patienten (spezielle Fragebögen sowie Interview);
- Untersuchung des Augenhintergrunds, um das Vorhandensein von Symptomen eines erhöhten Hirndrucks (Schwellung der Papille sowie Unschärfe ihrer Ränder) festzustellen;
- Um den Zustand des Schädels zu beurteilen, wird eine Röntgenaufnahme durchgeführt;
- Computer- und Magnetresonanztomographie des Kopfes, um die Struktur des Gehirns und des Schädels Schicht für Schicht zu untersuchen und das Vorhandensein von Symptomen einer vorzeitigen Fusion der Schädelnähte sowie zusätzlich eines Hydrozephalus (aufgrund des erhöhten Hirndrucks sammelt sich überschüssige Gehirn-Rückenmarks-Flüssigkeit an (dies ist die Gehirn-Rückenmarks-Flüssigkeit, die den Stoffwechselprozess sowie die Ernährung des Gehirns fördert)) festzustellen.
- Röntgenaufnahme der Füße und Hände, um die Ursache der Fingerversteifung zu ermitteln (dies ist wichtig für die Planung nachfolgender chirurgischer Eingriffe);
- Möglicherweise ist eine Konsultation mit einem medizinischen Genetiker und einem Neurochirurgen erforderlich.
Tests
Es werden genetische Analysen häufiger Mutationen im FGFR2-Typ-Gen durchgeführt.
Differenzialdiagnose
Dieses Syndrom muss von anderen genetischen Pathologien unterschieden werden, bei denen eine Kraniosynostose beobachtet wird. Dies sind Krankheiten wie das Pfeiffer-, Crouzon-, Saethre-Chotzen- und Carpenter-Syndrom. Molekulargenetische Testmethoden werden eingesetzt, um diese Anomalien auszuschließen.
Wen kann ich kontaktieren?
Behandlung Aper-Syndrom
Eine Operation gilt als die einzige wirksame Behandlung des Apert-Syndroms – sie hilft bei der Korrektur einzelner körperlicher Defekte und korrigiert auch eine geistige Behinderung.
Bei diesem Eingriff wird die Kronennaht verschlossen, um mögliche Hirnverletzungen zu verhindern. Die gängigste Technik ist die kraniofaziale Distraktion, bei der der Schädel schrittweise gezogen wird. Zur Beseitigung einzelner Gesichtsdefekte werden kieferorthopädische und/oder kieferorthopädische Operationen durchgeführt.
Darüber hinaus werden die Patienten einer chirurgischen Korrektur der Fingerversteifung unterzogen.
Verweise
Medizinische Genetik. Nationaler Führer. Herausgegeben von Ginter EK, Puzyrev VP GEOTAR-Media. 2022