Facharzt des Artikels

Neue Veröffentlichungen
Angelman-Syndrom bei Kindern und Erwachsenen
Zuletzt überprüft: 04.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.

Es gibt eine Reihe von Krankheiten, bei denen Ausdrücke wie „Pass auf dich auf, dann wirst du nicht krank“ zumindest lächerlich klingen. Dabei handelt es sich um Pathologien, bei denen dem Körper des Kindes bereits vor der Geburt einige geistige und körperliche Anomalien innewohnen, für die jedoch nicht die Eltern verantwortlich sind. Solche Krankheiten werden durch Mutationen oder Anomalien im Chromosomensatz verursacht und werden als chromosomal oder genetisch bedingt bezeichnet. Angelman-Syndrom, Down-Syndrom, Patau-Syndrom, Edwards-Syndrom, Turner-Syndrom, Prader-Willi-Syndrom – dies ist nur ein Teil der genetischen Erkrankungen aus einer recht ansehnlichen Liste.
Happy-Man-Syndrom
Dieses Mal sprechen wir über die Krankheit, die nach dem englischen Kinderarzt Harry Angelman benannt ist. Er sprach dieses Problem erstmals 1965 an, nachdem er am Vortag in seiner Praxis drei ungewöhnliche Kinder mit gemeinsamen, eigentümlichen Symptomen angetroffen hatte. Der Arzt nannte diese Kinder Puppenkinder und schrieb einen Artikel über sie, der zunächst „Marionettenkinder“ hieß. Der Artikel selbst und sein Titel wurden unter dem Eindruck eines Gemäldes verfasst, das in einem der Museen von Verona zu sehen war. Das Gemälde zeigte einen lachenden Jungen und trug den Titel „Der Puppenjunge“. Die Assoziation des auf dem Gemälde dargestellten Kindes mit den drei Kindern, die Angelman einst in seiner Praxis angetroffen hatte, veranlasste den Kinderarzt, die Kinder aufgrund ihrer Krankheit in einer Gruppe zusammenzufassen.
Es ist nicht verwunderlich, dass die im Artikel erwähnten Kinder von anderen Ärzten nicht beachtet wurden. Schließlich schienen sie auf den ersten Blick völlig unterschiedliche Krankheiten zu haben, so unterschiedlich war das allgemeine Krankheitsbild in drei verschiedenen Fällen. Vielleicht hätte die „neue“ Chromosomenpathologie andere Wissenschaftler interessiert, doch damals war die Genetik noch nicht weit genug entwickelt, um die Hypothese des englischen Arztes zu bestätigen. Daher wurde der Artikel nach einem gewissen Interesse lange Zeit in den Hintergrund gedrängt.
Die nächste Erwähnung des Angelman-Syndroms, wie der Artikel des englischen Kinderarztes G. Angelman nun hieß, stammt aus den frühen 80er Jahren des 20. Jahrhunderts. Und erst 1987 gelang es, den Grund herauszufinden, warum ein kleiner Teil der Kinder mit solchen Abweichungen geboren wird, dass sie von außen ständig lächeln und glücklich wirken. Tatsächlich stimmt dies überhaupt nicht, und das Lächeln ist nur eine Grimasse, hinter der sich eine unglückliche menschliche Seele und der Schmerz der Eltern verbergen.
Epidemiologie
Laut Statistik kann sich eine Chromosomenmutation bei einem Kind sowohl vor dem Hintergrund ähnlicher Mutationen bei den Eltern als auch ohne solche entwickeln. Es gibt keine eindeutige erbliche Natur des Angelman-Syndroms (AS), aber die Wahrscheinlichkeit, dass Eltern mit Chromosomenmutationen eine Pathologie entwickeln, ist recht hoch.
Interessant ist auch, dass, wenn in einer Familie bereits ein Kind mit AS lebt, die Wahrscheinlichkeit, dass ein zweites Kind die gleiche Störung hat, bei einem Prozent liegt, selbst wenn die Eltern gesund sind.
Es gibt noch keine genauen Statistiken über die Anzahl der Patienten mit Angelman-Syndrom. Der Grund dafür könnte die Vielfalt der Symptome sein, die in einer bestimmten Zusammensetzung auftreten oder lange Zeit überhaupt nicht auftreten können. Es wird angenommen, dass die Prävalenz der Krankheit bei 1 Kind pro 20.000 Neugeborenen liegt. Diese Zahl ist jedoch sehr ungefähr.
Ursachen Angelman-Syndrom
Das Angelman-Syndrom ist eine medizinische Bezeichnung für eine Chromosomenerkrankung, aber bei weitem nicht die einzige. Man nennt diese Krankheit Puppenkinder-Syndrom, Happy-Puppet-Syndrom, Petruschka-Syndrom und Lachpuppen-Syndrom. Man denkt sich alle möglichen Namen aus (manchmal sogar beleidigend für die Patienten selbst und ihre Eltern), aber eine Krankheit ist eine Krankheit, egal wie komisch sie aussieht und egal, was die Gründe dafür sind.
Und die Gründe für die Entwicklung des Angelman-Syndroms sind, wie bei vielen anderen genetischen Pathologien, in allen Fällen Störungen in der Struktur eines der Chromosomen oder des gesamten Chromosomensatzes. Aber in unserem Fall liegt das ganze Problem im Chromosom 15, das von der Mutter weitergegeben wird. Das heißt, das väterliche Chromosom weist in diesem Fall keine Abweichungen auf, das weibliche erfährt jedoch bestimmte Mutationen.
Je nach Art der Chromosomenanomalie wird das Angelman-Syndrom als Chromosomenmutation klassifiziert. Zu diesen Mutationen zählen:
- Eine Deletion (Fehlen eines Abschnitts eines Chromosoms, der einen bestimmten Satz von Genen enthält; wenn eines der Gene fehlt, sprechen wir von einer Mikrodeletion), die das Ergebnis von zwei Brüchen und einer Reunion ist, bei der ein Abschnitt des ursprünglichen Chromosoms verloren geht.
- Duplikation (das Vorhandensein eines zusätzlichen Abschnitts in einem Chromosom, der eine Kopie eines vorhandenen ist), was in den meisten Fällen zum Tod einer Person und seltener zu Unfruchtbarkeit führt.
- Inversion (Umkehrung eines der Chromosomenabschnitte um 180 Grad, d. h. in die entgegengesetzte Richtung, und dann befinden sich die Gene darin in der umgekehrten Reihenfolge), wenn die gebrochenen Enden des Chromosoms in einer anderen Reihenfolge als der ursprünglichen verbunden sind.
- Insertion (wenn ein Teil des genetischen Materials in einem Chromosom nicht an seinem Platz ist),
- Translokation (wenn ein bestimmter Abschnitt eines Chromosoms an ein anderes Chromosom angehängt wird; eine solche Mutation kann ohne Verlust von Abschnitten gegenseitig sein).
Wenn das Kind von einer ahnungslosen Mutter ein mutiertes Chromosom erhält, ist es dazu verdammt, mit Anomalien geboren zu werden. Als häufigste Ursache des Angelman-Syndroms gilt nach wie vor eine Deletion des mütterlichen 15. Chromosoms, bei der ein kleiner Abschnitt fehlt. Als seltenere Mutationen beim „Laughing Doll“-Syndrom gelten:
- Translokation,
- unipaternale Disomie (wenn das Kind ein Chromosomenpaar vom Vater erhalten hat, fehlt das mütterliche Chromosom),
- Mutation von Genen in der DNA, die sowohl das wichtigste (genetische) Baumaterial als auch Anweisungen für seine korrekte Verwendung sind (insbesondere Mutation des ube3a-Gens im mütterlichen Chromosom).
Das Vorhandensein einer dieser Mutationen bei den Eltern ist ein Risikofaktor für die Entwicklung des Angelman-Syndroms bei Kindern. Aber nicht nur Chromosomenmutationen, sondern auch Genommutationen (die mit einer quantitativen Veränderung des Chromosomensatzes verbunden sind und häufiger vorkommen als Chromosomenmutationen) können die Entwicklung der Krankheit bei einem Kind provozieren. Zu den häufigsten Genommutationen gehört die Chromosomentrisomie (wenn der Chromosomensatz einer Person mehr als 46 Chromosomen umfasst).
Damit bei einem Kind eine Pathologie auftritt, ist es überhaupt nicht notwendig, dass die Eltern Chromosomenanomalien haben. Dennoch gibt es einen gewissen Prozentsatz von Patienten, deren Krankheit erblich bedingt ist.
Pathogenese
Tauchen wir etwas tiefer in die Biologie, genauer gesagt in die Genetik, ein. Die genetische Information jedes einzelnen menschlichen Organismus ist in 23 Chromosomenpaaren enthalten. Ein Chromosom eines Paares wird vom Vater an das Kind weitergegeben, das andere von der Mutter. Alle Chromosomenpaare unterscheiden sich in Form und Größe und tragen bestimmte Informationen. So ist das 23. Chromosomenpaar (X- und Y-Chromosomen) für die Ausbildung der Geschlechtsmerkmale des Babys verantwortlich (XX – Mädchen, XY – Junge, während das Y-Chromosom nur vom Vater an das Kind weitergegeben werden kann).
Im Idealfall erhält ein Kind von seinen Eltern 46 Chromosomen, die seine genetischen Merkmale bilden und ihn als Individuum prägen. Eine größere Anzahl von Chromosomen wird als Trisomie bezeichnet und gilt als Abweichung von der Norm. Beispielsweise verursacht das Vorhandensein von Chromosom 47 im Chromosomensatz (Karyotyp, der Art und individuelle Merkmale bestimmt) das Auftreten des Down-Syndroms.
Wenn die Chromosomen mit einem speziellen Farbstoff angefärbt werden, sind unter dem Mikroskop Streifen unterschiedlicher Schattierungen erkennbar. In jedem Streifen befindet sich eine große Anzahl von Genen. Alle diese Streifen werden von Wissenschaftlern nummeriert und haben einen festen Standort. Das Fehlen eines Streifens gilt als Abweichung von der Norm. Beim Angelman-Syndrom fehlen sehr häufig Abschnitte des mütterlichen Chromosoms im Intervall q11-q13 im langen Arm, dessen Anzahl an DNA-Basen nur etwa 4 Millionen beträgt.
Der Hauptbestandteil des Chromosoms ist ein unglaublich langes DNA-Molekül mit Tausenden von Genen und Dutzenden und Hunderten von Millionen stickstoffhaltigen Basen. So enthält Chromosom 15, das für die Entstehung des Angelman-Syndroms und mehrerer anderer Erkrankungen verantwortlich ist, 1200 Gene und etwa 100 Millionen Basen. Jegliche Störungen in der Struktur des DNA-Moleküls wirken sich mit Sicherheit auf das Aussehen und die Entwicklung des ungeborenen Kindes aus.
Die in Genen enthaltene genetische Information wird in Proteine oder RNA umgewandelt. Dieser Prozess wird Genexpression genannt. Auf diese Weise erhält die von den Eltern erhaltene genetische Information Form und Inhalt, die in ihrem einzigartigen weiblichen oder männlichen Erben verkörpert werden.
Es gibt eine Reihe von Pathologien mit einem nicht-klassischen Vererbungstyp, darunter das Angelman-Syndrom, bei dem die von den Eltern als Teil gepaarter Chromosomen erhaltenen Gene einen einzigartigen Abdruck der Eltern tragen und sich auf unterschiedliche Weise manifestieren.
Das Angelman-Syndrom ist ein markantes Beispiel für genomische Prägung, bei der die Genexpression im Körper des Kindes direkt davon abhängt, von welchem Elternteil die Allele stammen (verschiedene Formen eines Gens, die von Vater und Mutter stammen und sich auf identischen Abschnitten gepaarter Chromosomen befinden). Das heißt, nur Anomalien im mütterlichen Chromosom führen zur Entwicklung des Syndroms, während Mutationen und strukturelle Störungen des väterlichen Chromosoms völlig andere Pathologien verursachen.
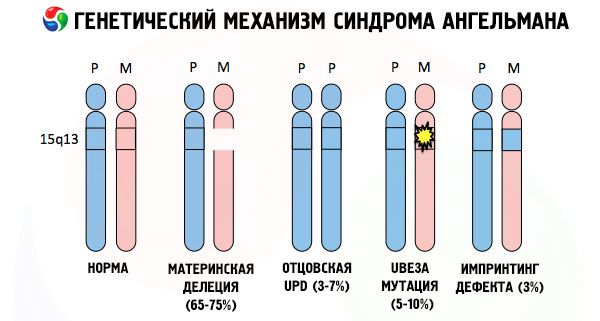
Bei dieser Erkrankung fehlen bestimmte Gene im mütterlichen Chromosom oder es kommt zu einem Verlust bzw. einer Verringerung der Aktivität einzelner Gene (in den meisten Fällen handelt es sich um das Gen ube3a, das am Stoffwechsel von Ubiquitin beteiligt ist, einem Protein, das den Abbau anderer Proteine reguliert). Infolgedessen werden bei dem Kind geistige Entwicklungsstörungen und körperliche Missbildungen diagnostiziert.
Symptome Angelman-Syndrom
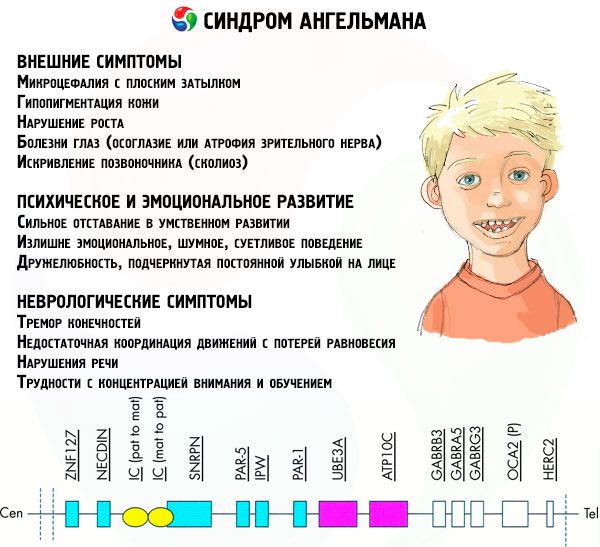
Die Symptome des Angelman-Syndroms beeinflussen verschiedene Aspekte des Lebens und der Entwicklung eines Kindes: körperlich, neurologisch und geistig. Auf dieser Grundlage lassen sich drei Symptomgruppen identifizieren, die auf die Entwicklung dieser Pathologie hinweisen.
- Äußere oder körperliche Symptome:
- ein im Verhältnis zum Körper und den Gliedmaßen normaler Größe unverhältnismäßig kleiner Kopf,
- zu breiter Mund,
- es ist fast immer ein Lächeln im Gesicht (mit offenem Mund),
- spärliche Zähne,
- schmale Oberlippe,
- häufig heraushängende breite Zunge,
- vorstehender Unterkiefer,
- spitzes Kinn,
- sehr helle Haut, oft Haare (Albinismus, verbunden mit der Tatsache, dass der Körper das Pigment Melanin nicht produziert),
- dunkle Flecken auf heller Haut (Hypopigmentierung aufgrund unzureichender Melaninproduktion)
- körperliche oder äußere Symptome: Augenerkrankungen wie Strabismus oder Sehnervenatrophie,
- Verkrümmung der Wirbelsäule (Skoliose),
- steife Beine (beim Gehen beugt eine Person ihre Beine aufgrund der geringen Beweglichkeit der Gelenke nicht an den Knien, daher der Vergleich mit dem Gang einer Puppe).
- Symptome im Zusammenhang mit der geistigen und emotionalen Entwicklung:
- schwere geistige Behinderung,
- übermäßig emotionales, lautes, pingeliges Verhalten,
- häufiges Händeklatschen,
- ausgedrückte Freundlichkeit, unterstrichen durch ein ständiges Lächeln im Gesicht,
- häufiges Lachen ohne Grund.
- Neurologische Symptome:
- Zittern der Gliedmaßen,
- unzureichende Bewegungskoordination mit Gleichgewichtsverlust,
- verminderter Muskeltonus,
- verschiedene Schlafstörungen,
- häufige hysterische Anfälle in der Kindheit,
- Sprachstörungen (das Kind beginnt spät zu sprechen, hat schlechte Kommunikationsfähigkeiten und spricht undeutlich),
- Hyperaktivität vor dem Hintergrund erhöhter Erregbarkeit,
- Konzentrations- und Lernschwierigkeiten.
Dies ist jedoch ein verallgemeinertes Bild der Krankheit. Tatsächlich hängt das klinische Bild des Angelman-Syndroms weitgehend vom Entwicklungsstadium der Krankheit und der Art der Chromosomenmutation ab, die die Pathologie verursacht hat. Dies bedeutet, dass die Krankheitssymptome bei verschiedenen Patienten erheblich unterschiedlich sein können, was es lange Zeit nicht ermöglichte, die Pathologie von anderen mit ähnlichem Krankheitsbild zu unterscheiden.
Unter der Gesamtzahl der Symptome können wir diejenigen hervorheben, die ausnahmslos für alle Patienten charakteristisch sind:
- schwere geistige Behinderung,
- unangemessenes Verhalten (unangemessenes Lachen, erhöhte Erregbarkeit, Konzentrationsschwäche, Euphoriezustand),
- Unterentwicklung der motorischen Fähigkeiten,
- schlechte Bewegungskoordination, Gangataxie (ungleichmäßiges Tempo, Schwanken von einer Seite zur anderen usw.), Zittern der Gliedmaßen.
- Sprachentwicklungsstörung mit einem Überwiegen nonverbaler Kommunikationsmittel.
Zu den Symptomen, mit denen die überwiegende Mehrheit der Patienten konfrontiert ist, zählen:
- Missverhältnis zwischen Kopf und Körper aufgrund einer verzögerten körperlichen Entwicklung,
- Bei vielen Patienten ist die Schädelform so, dass die Größe des Gehirns kleiner bleibt als bei gesunden Menschen (Mikrozephalie),
- epileptische Anfälle vor dem dritten Lebensjahr mit fortschreitender Abnahme der Stärke und Häufigkeit im höheren Alter,
- Verzerrung der EEG-Parameter (Schwankungen und hohe Amplitude niederfrequenter Wellen).
Diese Symptome kommen recht häufig vor, bei 20 % der Patienten mit Angelman-Syndrom treten sie jedoch nicht auf.
Noch seltener können folgende Krankheitserscheinungen diagnostiziert werden:
- starkes oder leichtes Schielen,
- schlechte Kontrolle der Zungenbewegung, was dazu führt, dass Patienten oft ohne Grund die Zunge herausstrecken,
- Schluck- und Saugbeschwerden, insbesondere bei Kleinkindern,
- Störung der Haut- und Augenpigmentierung,
- beim Gehen erhobene oder gebeugte Arme,
- Hyperreflexie,
- Schlafstörungen, insbesondere im Kindesalter,
- häufiger Speichelfluss,
- unstillbarer Durst,
- übermäßig aktive Kaubewegungen,
- Überempfindlichkeit gegen Hitze,
- flacher Hinterkopf,
- vorstehender Unterkiefer,
- glatte Handflächen.
Ein großer Prozentsatz der Patienten leidet unter Problemen beim Wasserlassen, die sie schlecht kontrollieren können, einer eingeschränkten Feinmotorik, die zu Schwierigkeiten bei der Selbstversorgung und beim Lernen führt, sowie an Übergewicht. Fast alle Patienten kommen später in die Pubertät als gesunde Altersgenossen.
Kinder mit Angelman-Syndrom nehmen mündliche Sprache gut wahr und verstehen sie, möchten sich aber nicht an Gesprächen beteiligen und beschränken ihre Sprache auf einige Dutzend Wörter, die im Alltag notwendig sind. Im Erwachsenenalter sehen solche Patienten jedoch jünger aus als ihre Altersgenossen ohne genetische Pathologie.
Viele Symptome des Angelman-Syndroms sind unbeständig, sodass sich das Krankheitsbild mit zunehmendem Alter deutlich verändert. Krämpfe und epileptische Anfälle werden seltener oder verschwinden ganz, der Patient wird weniger erregbar und der Schlaf verbessert sich.
Komplikationen und Konsequenzen
Das Angelman-Syndrom ist eine schwere, derzeit nahezu unheilbare Chromosomenerkrankung, die den Patienten die Möglichkeit eines normalen Lebens nimmt. Wie das Leben eines Kindes mit AS aussehen wird, hängt maßgeblich von der Art der Chromosomenanomalie ab.
Die Verdoppelung eines Chromosomenabschnitts ist in den meisten Fällen mit dem Leben unvereinbar. Und selbst wenn solche Patienten nicht im Säuglingsalter sterben und die Pubertät erreichen, haben sie keine Chance, Kinder zu bekommen.
Die Deletion oder das Fehlen eines Teils der Gene, die beim Angelman-Syndrom am häufigsten auftreten, behindert das Kind beim Laufen- und Sprechenlernen. Solche Kinder haben eine schwerere Form der geistigen Behinderung, epileptische Anfälle treten häufiger auf und sind deutlich intensiver als bei Patienten mit anderen Chromosomenanomalien.
Liegt nur eine Mutation eines Gens vor, können dem Kind mit der entsprechenden Aufmerksamkeit und Herangehensweise die Grundlagen der Selbstfürsorge, Kommunikation und Interaktion in einer Gruppe beigebracht werden, obwohl es in seiner Entwicklung immer noch hinter seinen Altersgenossen zurückbleibt.
Für Kinder mit Angelman-Syndrom, die von Natur aus freundlich sind, ist die Liebe und Aufmerksamkeit ihrer Eltern das Wichtigste. Nur dann trägt die Erziehung des Kindes Früchte, auch wenn sie nur gering ist. Patienten mit AS können natürlich nicht in einer Regelschule lernen. Sie benötigen spezielle Kurse, in denen den Kindern zunächst Konzentration beigebracht und dann schrittweise die Grundlagen des Schulwissens vermittelt werden.
Diagnose Angelman-Syndrom
Das Angelman-Syndrom ist eine angeborene Entwicklungsstörung. Aufgrund bestimmter Umstände ist es jedoch oft unmöglich, es im Säuglings- und Kleinkindalter zu diagnostizieren. Dies liegt an der Unspezifität und der schwachen Ausprägung der Symptome bei Säuglingen und Kindern unter 3 Jahren. Und die Prävalenz der Krankheit in unserem Land ist nicht so groß, dass Ärzte gelernt hätten, sie unter ihresgleichen zu erkennen.
Das Angelman-Syndrom bei Säuglingen kann sich in einem verminderten Muskeltonus äußern, der sich in Problemen beim Füttern (Schwäche des Saug- und Schluckreflexes) und späteren Schwierigkeiten beim Laufenlernen äußert (solche Kinder beginnen viel später zu laufen). Diese Symptome sind die ersten Anzeichen einer Entwicklungsstörung beim Baby, die durchaus mit einer Chromosomenanomalie verbunden sein kann. Nur eine genetische Analyse kann diese Annahme bestätigen.
Besonderes Augenmerk wird auf Kinder gelegt, deren Eltern an verschiedenen Genom- oder Chromosomenstörungen leiden. Schließlich kann sich die Krankheit zunächst nicht manifestieren. Wird die Pathologie jedoch rechtzeitig erkannt, kann durch intensive Zusammenarbeit mit dem Kind ein deutlich höherer Lernerfolg erzielt und das Fortschreiten der Krankheit verlangsamt werden.
Liegen bei den Eltern verschiedene Chromosomenanomalien vor, wird bereits vor der Geburt des Babys eine genetische Analyse durchgeführt, da SA zu den Pathologien gehört, die bereits im Embryonalstadium festgestellt werden können.
Die Sammlung von Material für die genetische Forschung kann auf zwei Arten erfolgen:
- invasiv (mit einem gewissen Risiko, da zur Entnahme einer Fruchtwasserprobe ein Eingriff in die Gebärmutter erforderlich ist),
- nicht-invasiv (Analyse der DNA des Babys aus dem Blut der Mutter).
Anschließend werden folgende Untersuchungen durchgeführt:
- Fluoreszenz-in-situ-Hybridisierung (FISH-Methode) – Bindung einer mit einem speziellen Farbstoff markierten DNA-Sonde an die zu untersuchende DNA und anschließende Untersuchung unter dem Mikroskop.
- Analyse von Mutationen im ube3a-Gen und geprägten Genen,
- DNA-Methylierungsanalyse mit speziellen Methoden der Genetik.
Genetische Tests liefern bei Chromosomenanomalien relativ genaue Informationen, sodass werdende Eltern im Voraus wissen, worauf sie sich einstellen müssen. Es gibt jedoch Ausnahmen. Bei einer bestimmten Patientengruppe bleiben die Testergebnisse bei Vorliegen aller pathologischen Symptome normal. Das heißt, eine Pathologie kann nur durch sorgfältige Beobachtung des Kindes von frühester Kindheit an erkannt werden: wie es isst, wann es zu laufen und zu sprechen begann, ob es beim Gehen die Beine beugt usw.
Zu den instrumentellen Diagnosemethoden des Angelman-Syndroms zählen neben der FISH-Methode die Tomographie (CT oder MRT), die hilft, den Zustand und die Größe des Gehirns zu bestimmen, und ein Elektroenzephalogramm (EEG), das zeigt, wie einzelne Teile des Gehirns funktionieren.
Die endgültige Diagnose stellen Ärzte in der Regel im Alter von 3–7 Jahren, wenn der Patient bereits die meisten Symptome aufweist und die Dynamik der Krankheitsentwicklung sichtbar ist.
Welche Tests werden benötigt?
Differenzialdiagnose
Das Angelman-Syndrom ist eine genetische Erkrankung, die praktisch keine spezifischen Ausprägungen aufweist. Die meisten Symptome können gleichermaßen auf AS und andere genetische Erkrankungen hinweisen.
Die Differentialdiagnose des Angelman-Syndroms wird bei folgenden Pathologien durchgeführt:
- Pitt-Hopkins-Syndrom (Patienten zeichnen sich durch geistige Behinderung, fröhlichen Charakter, Lächeln, einen ziemlich großen und breiten Mund und Mikrozephalie aus). Der Unterschied besteht in Anfällen von Hyperventilation und Atemanhalten im Wachzustand.
- Christianson-Syndrom (Patienten sind geistig behinderte Menschen mit fröhlichem Gemüt, die nicht sprechen können und durch Mikrozephalie, Ataxie, Krämpfe und unwillkürliche Muskelbewegungen gekennzeichnet sind).
- Mowat-Wilson-Syndrom (Symptome: geistige Behinderung, epileptische Anfälle, spitzes Kinn, offener Mund, fröhlicher Gesichtsausdruck, Mikrozephalie). Besonderheiten: großer Augenabstand, nach innen geneigte Augen, abgerundete Nasenspitze, nach hinten gedrehte Ohrmuschel.
- Kabuki-Syndrom (gekennzeichnet durch leichte bis mittelschwere geistige Behinderung, Sprach- und Bewegungsstörungen, Muskelschwäche, epileptische Anfälle, Mikrozephalie, lange Intervalle zwischen Juckreiz und Koordinationsstörungen). Charakteristisch sind hochgezogene Augenbrauen, ein nach außen gerichteter seitlicher Teil des Unterlids, weit auseinanderstehende Augen, lange Lidspalten mit langen, dichten Wimpern.
- Rett-Syndrom (Differenzierung zum AS bei Frauen). Symptome: verzögerte Sprachentwicklung, Krampfanfälle, Mikrozephalie. Der Unterschied besteht darin, dass der Gesichtsausdruck fehlt und Apnoe- und Apraxie-Anfälle auftreten, die sich mit der Zeit verschlimmern.
- Autosomal-rezessives Mental-Tardation-Syndrom 38 (Symptome: ausgeprägte geistige Behinderung mit Verzögerungen in Motorik und Sprache, Muskelschwäche, Ernährungsprobleme im Säuglingsalter, Impulsivität). Charakteristisches Merkmal ist die blaue Farbe der Iris.
- MECP-2-Genduplikationssyndrom (Differenzierung zur SA bei Männern). Symptome: schwere geistige Behinderung, Muskelschwäche seit der Kindheit, Sprachstörungen oder Sprachlosigkeit, Epilepsie. Besonderheiten: progressive Myopathie, ständig wiederkehrende Infektionen.
- Kleefstra-Syndrom (Symptome: Sprach- und Denkstörungen, Muskelschwäche, Schlafstörungen, Unaufmerksamkeit, offener Mund, Hyperaktivität, Krampfanfälle, Ataxie, Gleichgewichtsstörungen). Charakteristische Merkmale: flaches Gesicht, kurze Stupsnase, weit auseinanderstehende Augen, große, nach oben gezogene Unterlippe, aggressive Ausbrüche.
- Smith-Magenis-Syndrom (gekennzeichnet durch Krampfanfälle, Schlafstörungen, Störungen der geistigen und motorischen Entwicklung). Charakteristische Merkmale sind ein breites und flaches Gesicht und eine markante Stirn.
- Koolen-de-Vries-Syndrom (leichte bis mittelschwere geistige Behinderung, Muskelschwäche, Krampfanfälle, Freundlichkeit). Besondere Merkmale: langes Gesicht mit hoher Stirn, abstehende Ohren, schräg stehende Augen, hohe Gelenkbeweglichkeit, angeborene Herzfehler.
- Phelan-McDermid-Syndrom (Symptome: geistige Behinderung, Sprachstörungen oder Sprachlosigkeit). Besonderheiten: große Hände mit ausgeprägter Muskulatur, angeborene Muskelschwäche, schwaches Schwitzen.
Pathologien wie Adenylsuccinatmangel, autosomal-rezessives mentales Retardierungssyndrom 1, Chromosom 2q23.1-Duplikationssyndrom, FOXG1-, STXBP1- oder MEF2C-Gen-Haploinsuffizienz-Syndrome und einige andere können mit Symptomen „prahlen“, die dem Angelman-Syndrom ähnlich sind.
Die Aufgabe des Arztes besteht darin, eine genaue Diagnose zu stellen, das Angelman-Syndrom von Erkrankungen mit ähnlichen Symptomen zu unterscheiden und eine wirksame Behandlung zu verschreiben, die dem diagnostizierten Stadium der Krankheit entspricht.
Wen kann ich kontaktieren?
Behandlung Angelman-Syndrom
Das Angelman-Syndrom ist eine jener Erkrankungen, für die die Medizin noch immer nach einer wirksamen Behandlung sucht. Die ätiologische Behandlung der Krankheit befindet sich in der Entwicklungsphase verschiedener Methoden und Mittel, von denen viele noch nicht am Menschen erprobt sind. Das bedeutet, dass sich Ärzte vorerst auf eine symptomatische Therapie beschränken müssen, die dazu beiträgt, die beneidenswerte Situation von Kindern und Erwachsenen mit Marionettensyndrom, die unter epileptischen Anfällen, Speichelfluss, Hypotonie und Schlafstörungen leiden, etwas zu lindern.
So ist es möglich, die Häufigkeit und Stärke epileptischer Anfälle mithilfe eines richtig ausgewählten Antikonvulsivums zu reduzieren. Die ganze Schwierigkeit besteht jedoch darin, dass sich Anfälle bei Patienten mit SA von gewöhnlichen epileptischen Anfällen dadurch unterscheiden, dass sie durch mehrere Anfallsarten gekennzeichnet sind, was bedeutet, dass der Zustand durch die gleichzeitige Verabreichung mehrerer Medikamente gelindert werden kann.
Die gängigsten Antikonvulsiva zur Behandlung des Angelman-Syndroms sind: Valproinsäure, Topiramat, Lamotrigin, Levetiracetam, Clonazepam und darauf basierende Medikamente. Seltener werden Medikamente auf Basis von Carmazepin, Phenytoin, Phenobarbital und Ethosuximid eingesetzt, da einige von ihnen eine paradoxe Wirkung haben können, die in der Verstärkung und Häufigkeit epileptischer Anfälle besteht. Dies ist der Fall, wenn das Medikament im Rahmen einer Monotherapie angewendet wird.
Zur Behandlung von Speichelfluss werden üblicherweise zwei Methoden eingesetzt: medikamentös (mit Medikamenten, die die Speichelproduktion unterdrücken) und chirurgisch, wobei die Speichelgänge replantiert werden. Bei SA gelten diese Methoden jedoch als unwirksam, und die Frage bleibt ungeklärt. Eltern und Betreuer solcher Patienten müssen diesem Problem besondere Aufmerksamkeit schenken, da die Patienten ihren Speichelfluss meist nicht selbst kontrollieren können und manche einfach nicht in der Lage sind, für sich selbst zu sorgen.
Ein weiteres Problem ist die kurze Schlafdauer. Kinder mit Angelman-Syndrom schlafen oft nicht länger als 5 Stunden, was sich negativ auf die Funktion des gesamten Körpers auswirkt. Leicht erregbare, aktive Kinder, die gerne spielen und kommunizieren (auch wenn sie versuchen, sich auf nonverbale Methoden zu beschränken), sind tagsüber merklich müde. Um sich gut auszuruhen, braucht der Körper tiefen, vollen Schlaf, aber genau hier liegt der Haken.
Es scheint, dass Beruhigungsmittel (Phenothiazine und atypische Antipsychotika), die das Nervensystem beruhigen, ausreichen sollten, um den Schlaf bei erregbaren Patienten zu verbessern. Bei AS ist die Einnahme solcher Medikamente jedoch mit negativen Auswirkungen verbunden. Daher bevorzugen Ärzte nach wie vor milde Schlaftabletten wie Melatonin (ein natürliches Hormonpräparat auf Basis des Schlafhormons), das den Patienten eine Stunde vor dem Schlafengehen in einer Menge von 1 Tablette verabreicht wird, und Diphenhydramin. Die Häufigkeit der Verabreichung und die Dosierung werden vom Arzt je nach Zustand und Alter des Patienten festgelegt.
Patienten mit Angelman-Syndrom haben manchmal Probleme mit der Verdauung und dem Stuhlgang. Abführmittel (vorzugsweise pflanzliche) können den Stuhlgang verbessern.
Oder Sie können das Problem anders angehen, wie es amerikanische Ärzte taten, basierend auf einigen Methoden zur Behandlung von Autismus, da viele für AS charakteristische Symptome auch für Autismus charakteristisch sind (Impulsivität, unwillkürliche Bewegungen, sich wiederholende Handlungen, Aufmerksamkeitsdefizit, Kommunikationsprobleme usw.). Es wurde festgestellt, dass sich die Einführung des Hormons Sekretin, das die Verdauung und den Stuhlgang normalisiert, positiv auf die Aufmerksamkeit der Patienten auswirkt und Oxytocin dazu beiträgt, die kognitiven Fähigkeiten und das Gedächtnis des Kindes zu verbessern und das Verhalten zu korrigieren.
Zwar reichen Hormone allein nicht aus, insbesondere bei Kindern. Beim Angelman-Syndrom sind Verhaltenstherapie, die Zusammenarbeit mit einem Psychologen und einem Logopäden (Unterricht in nonverbaler Kommunikation und Gebärdensprache) angezeigt. Die Erziehung solcher Kinder sollte auf einem individuellen Programm basieren, an dem speziell ausgebildete Lehrer, ein Psychologe und Eltern teilnehmen. Leider ist dies nicht überall möglich, und Familien werden mit ihrem Problem allein gelassen.
Da viele junge Patienten mit AS unter Muskelschwäche und Gelenkproblemen leiden, wird der Physiotherapie große Aufmerksamkeit geschenkt. Am häufigsten greifen Ärzte auf Paraffinanwendungen, Elektrophorese und Magnetfeldtherapie zurück.
Aktive Tonikummassagen und spezielle Übungen des therapeutischen Körpertrainings helfen dem kranken Kind nach einiger Zeit, auf den Beinen zu stehen und sicher zu gehen. Besonders hilfreich ist in diesem Zusammenhang Aquagymnastik, die bei SA im kühlen Wasser empfohlen wird. Sie stärkt den Muskeltonus und lehrt das Kind, seinen Körper zu kontrollieren und Bewegungen zu koordinieren.
Antikonvulsive Behandlung
Das gefährlichste Symptom des Angelman-Syndroms sind epilepsieähnliche Anfälle. Dieses Symptom tritt bei 80 % der Patienten auf, was bedeutet, dass allen Patienten eine wirksame antikonvulsive Behandlung verschrieben werden muss.
Die Behandlung epileptischer Anfälle erfolgt mit Hilfe von Vitaminen und Antikonvulsiva. Beim Angelman-Syndrom, das von einem Krampfsyndrom begleitet wird, sind Vitamine der Gruppe B sowie die Vitamine C, D und E hilfreich. In diesem Fall ist es jedoch sehr gefährlich, eine Vitamintherapie selbst zu verschreiben, da eine unkontrollierte Einnahme von Vitaminen die Wirksamkeit von Antiepileptika verringern und neue, schwerere und länger anhaltende Anfälle hervorrufen kann.
Die Auswahl der Antiepileptika und die Verschreibung ihrer wirksamen Dosierung sollten ebenfalls von einem Facharzt vorgenommen werden. Er entscheidet auch, ob ein Medikament ausreicht oder ob der Patient über einen längeren Zeitraum zwei oder mehr Medikamente einnehmen muss.
Den meisten Patienten verschreiben Ärzte Valproinsäure-Medikamente (Valproinsäure, Depakine, Convulex, Valparin usw.), die Krampfanfälle verhindern und die Stimmung und den Geisteszustand der Patienten verbessern.
Valproinsäure ist in Form von Tabletten, Sirup und Injektionslösungen erhältlich. Das beliebteste Medikament ist das Retardpräparat „Depakine“ in Tablettenform und als Lösung zur intravenösen Verabreichung. Die Dosierung des Arzneimittels wird vom Arzt individuell in Abhängigkeit von Gewicht, Alter und Zustand des Patienten festgelegt.
Das Medikament wird 2 bis 3 Mal täglich zu den Mahlzeiten eingenommen. Die durchschnittliche Tagesdosis beträgt 20–30 mg pro Kilogramm Körpergewicht des Patienten, die maximale Dosis beträgt 50 mg/kg pro Tag.
Kontraindikationen für die Anwendung. Nicht anwenden bei Leber- und Pankreasfunktionsstörungen, hämorrhagischer Diathese, Hepatitis, Porphyrie und Überempfindlichkeit gegen das Arzneimittel.
Zu den Nebenwirkungen zählen Handzittern, Verdauungs- und Stuhlstörungen sowie Veränderungen des Körpergewichts.
Auch bei SA ist „Topiramat“ ein Mittel der Wahl. Es wird in Tablettenform hergestellt und sowohl als Teil einer Monotherapie als auch in Kombination mit anderen Arzneimitteln eingesetzt.
Art der Anwendung und Dosierung. Nehmen Sie die Tabletten unabhängig von der Nahrungsaufnahme oral ein. Die anfängliche Tagesdosis für Erwachsene beträgt 25–50 mg, für Kinder 0,5–1 mg/kg. Jede Woche wird die Dosierung gemäß den Anweisungen des Arztes erhöht.
Das Medikament sollte während der Schwangerschaft und Stillzeit sowie bei Überempfindlichkeit gegen seine Bestandteile nicht eingenommen werden. Das Medikament hat viele verschiedene Nebenwirkungen.
Medikamente, die ein Arzt bei Angelman-Syndrom verschreiben kann: Clomazepam, Rivotril, Lamotrigin, Seizar, Lamictal, Levetiracetam, Keppra, Epiterra usw.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Traditionelle Medizin und Homöopathie
Traditionelle Medikamente wie homöopathische Präparate sind natürlich relativ sicher, die Wirksamkeit einer solchen Behandlung des Angelman-Syndroms kann jedoch als umstritten angesehen werden.
Obwohl Volksheilmittel in manchen Fällen helfen können. Es geht um die Beendigung epileptischer Anfälle. In dieser Hinsicht kann eine Kräuterbehandlung sehr wirksam sein.
Eine gute Wirkung erzielt eine Arzneimittelkollektion auf Basis von Pfingstrose, Süßholz und Wasserlinse (die Komponenten werden in gleichen Mengen eingenommen). Die Kräuter müssen zu Mehl gemahlen werden. Bereits 2 Wochen nach Einnahmebeginn ist eine deutliche Abnahme der Anfallshäufigkeit festzustellen.
Lavendelabkochung (1 Teelöffel pro Glas kochendes Wasser) ist auch bei Krämpfen hilfreich. Die Mischung wird 5 Minuten gekocht und eine halbe Stunde lang ziehen gelassen. Das Arzneimittel wird 14 Tage lang abends eingenommen.
Ein wässriger (oder alkoholischer) Aufguss aus Herzgespann gilt als wirksam bei epileptischen Anfällen.
Von den homöopathischen Präparaten zur Vorbeugung von Anfällen beim Angelman-Syndrom können Sie Arzneimittel auf Basis von Kamille und Herzgespann, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum und Arsenicum album verwenden. Es sollte jedoch berücksichtigt werden, dass nur ein homöopathischer Arzt im Einzelfall wirksame und sichere Dosierungen von Arzneimitteln verschreiben kann.
Verhütung
Wie der Leser wahrscheinlich bereits verstanden hat, ist die Medizin jedoch noch nicht in der Lage, Genmutationen und andere Chromosomenanomalien zu verhindern oder zu korrigieren. Dies kann jedem passieren, da Kinder mit Angelman-Syndrom von gesunden Eltern geboren werden und die Genetik, die derzeit zu den am wenigsten erforschten Zweigen der Medizin zählt, dies noch nicht erklären kann.
Das Einzige, was man tun kann, ist, verantwortungsvoll mit der Schwangerschaftsplanung umzugehen, sich rechtzeitig anzumelden und Untersuchungen durchführen zu lassen. Aber auch hier gilt, dass eine solche Maßnahme eher pädagogischer als präventiver Natur ist, wie jede Untersuchung. Junge Eltern wissen jedoch im Voraus, worauf sie sich vorbereiten müssen, und entscheiden im Falle einer positiven Antwort, ob sie die Verantwortung für die Erziehung eines kranken Kindes übernehmen können.
Prognose
Die Prognose des Angelman-Syndroms hängt von der Art der Chromosomenanomalie und der Rechtzeitigkeit ihrer Erkennung ab. Am stärksten betroffen sind Kinder, deren Chromosom 15 Genlücken (Deletionen) aufweist. Die Wahrscheinlichkeit, dass solche Patienten gehen und sprechen können, ist äußerst gering. Andere Fälle können mit einem sorgfältigen Vorgehen und Liebe zu Ihrem Kind korrigiert werden.
Leider können solche Patienten keine vollwertigen Mitglieder der Gesellschaft werden, obwohl sie alles andere als dumm sind und Sprache und ihre Bedeutung verstehen. Sie werden jedoch ihr Leben lang Kommunikationsprobleme haben. Patienten können von Kindheit an Gebärdensprache lernen, aber sie können nicht gezwungen werden, mit Worten zu kommunizieren. Der Wortschatz „sprechender“ Patienten beschränkt sich auf das Minimum an Wörtern, die im Alltag verwendet werden (5-15 Wörter).
Was die Lebenserwartung und den allgemeinen Gesundheitszustand von Patienten mit Angelman-Syndrom betrifft, schwanken die Zahlen hier um Durchschnittswerte. Im Erwachsenenalter leiden die Patienten meist unter gesundheitlichen Problemen wie Skoliose und Fettleibigkeit, die bei richtiger Behandlung nicht lebensbedrohlich sind.