Facharzt des Artikels

Neue Veröffentlichungen
Gliom des Gehirns
Zuletzt überprüft: 29.06.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.
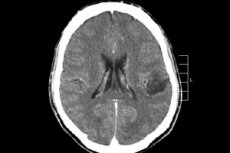
Unter den vielen Tumorprozessen des Zentralnervensystems wird am häufigsten ein Hirngliom diagnostiziert – dieser Begriff ist kollektiv, die Neoplasie umfasst alle diffusen oligodendroglialen und astrozytischen Herde, Astrozytome, Astroblastome usw. Ein solcher Tumor kann unterschiedlich bösartig sein und wird aus Gliastrukturen gebildet – Zellen, die um Neuronen herum lokalisiert sind. Gliome treten hauptsächlich in den Großhirnhemisphären, den Wänden der Hirnventrikel und dem Chiasma auf – dem Bereich der teilweisen Kreuzung der Sehnervenfasern. Äußerlich ist der Tumor ein knotiges Element von rosa oder rötlicher Tönung, runder oder spindelförmiger Konfiguration mit undeutlichen Grenzen. [ 1 ]
Epidemiologie
In etwa 5 % der Fälle sind Gliome mit erblichen Erkrankungen assoziiert – insbesondere mit Neurofibromatose und anderen Syndromen mit dominanter Vererbung. Experten weisen darauf hin, dass sich die überwiegende Mehrheit der Hirngliome sporadisch entwickelt, also ohne eindeutige Ursache.
Insgesamt machen primäre Neoplasien des Zentralnervensystems etwa 2 % aller Tumoren aus, das sind etwas mehr als 21 Fälle pro 100.000 Einwohner. Darunter sind Gliome in 35–36 % der Fälle, und mehr als 15 % davon sind Glioblastome.
Einigen Daten zufolge sind Männer häufiger von Gliomen betroffen als Frauen – besonders häufig tritt der Tumor bei Menschen über 50 Jahren auf.
Die weltweite Häufigkeit von Gliomen bei älteren Menschen hat in den letzten Jahrzehnten deutlich zugenommen. Die Gründe dafür sind noch nicht geklärt.
Nach der Definition der Weltgesundheitsorganisation wurden drei Hauptvarianten von Gliatomen identifiziert, die sich in ihren histologischen Merkmalen unterscheiden. Dies sind Oligodendrogliome, Astrozytome und kombinierte Oligoastrozytome. Die Inzidenz jedes Subtyps einer niedrig malignen Pathologie wurde nicht zuverlässig bestimmt. Einige Studien deuten auf einen Anstieg der Inzidenz von Oligodendrogliomen von 5 % auf 30 % und einen Rückgang der Inzidenz von Astrozytomen hin.
Gliale Tumoren können in das Hirngewebe eindringen und die überwiegende Mehrheit der niedrigmalignen Herde entartet innerhalb weniger Jahre. [ 2 ]
Ursachen Hirngliome
Hirngliome sind eine Gruppe von Tumoren, deren gemeinsames Merkmal ihre Entstehung aus Gliastrukturen des ZNS im Hirngewebe ist. Histologisch werden solche Tumoren in zwei Varianten unterteilt: hochmaligne und niedrigmaligne Gliome.
Die Quelle der Wucherungen sind Neurogliazellen (Astrozyten, Oligodendrozyten), die die strukturelle Grundlage und Lebensfähigkeit der Gehirnneuronen bilden.
Gliale Tumorprozesse unterscheiden sich erheblich in ihrer Struktur, ihren Mutationen in Genen, ihrer Aggressivität, ihren klinischen Merkmalen, ihren diagnostischen Eigenschaften, ihrem Ansprechen auf die Behandlung und ihrer Prognose. Embryonale und ependymale Neoplasien des Zentralnervensystems – insbesondere Medulloblastome und Ependymome – unterscheiden sich in ihrer histologischen Struktur, sind aber in Bezug auf die Behandlung ähnlich.
Gliaelemente wurden erstmals im späten 19. Jahrhundert als separate Strukturkategorie des Nervensystems klassifiziert.
Neurogliagewebe besteht aus Zellen mit Hilfsfunktionen: trophische, unterstützende, schützende und sekretorische. Neuronen und Gliozyten existieren zusammen, bilden zusammen das Nervensystem und sind für die allgemeinen Prozesse der Lebensaktivität des Organismus von großer Bedeutung.
Gliozyten werden grob in mehrere Hauptformen eingeteilt: Astrozyten, Oligodendrozyten, Ependymzellen und Mikroglia.
Bis heute können Wissenschaftler die Frage nach den zuverlässigen Ursachen der Entstehung neuroglialer Tumore nicht beantworten. Vermutlich tragen radioaktive Einflüsse, Infektionskrankheiten und Vergiftungen (insbesondere chemische und berufliche) negativ dazu bei. Auch der erbliche Faktor ist wichtig.
Hirngliome entstehen aus abnormen Neurogliozyten, die einen genetischen Defekt aufweisen, der zu abnormalem Wachstum und abnormaler Funktionalität führt – solche Strukturen werden als „unreif“ bezeichnet. Unvollständige Zellen befinden sich häufiger in einem Bereich, in dem sich der Tumor bildet.
Vereinfacht ausgedrückt ist die Gliabildung das Ergebnis chaotischen und sporadischen Wachstums modifizierter Neurogliazellen. Der Prozess kann sich aus Ependymozyten, Oligodendrozyten und Astrozyten (Astrozytom, einschließlich Riesenzellen und anaplastischem) entwickeln. [ 3 ]
Risikofaktoren
Obwohl Experten die Ursachen für die Entstehung glialer Onkopathologien nicht genau charakterisieren können, kann ihr Auftreten in einigen Fällen durch die Beseitigung der Hauptrisikofaktoren verhindert werden:
- Die Exposition gegenüber ionisierender Strahlung hat krebserregende Wirkungen und kann die Entwicklung von Leukämie und die Bildung von dichten Krebsprozessen, auch bei jungen Menschen, verursachen. Häufige und unangemessene radiologische medizinische Verfahren sowie ultraviolette Strahlung (einschließlich Solarium) haben ebenfalls potenzielle krebserregende Wirkungen und können Tumoren in verschiedenen Organen, einschließlich des Gehirns, verursachen.
- Berufsbedingte Nebenwirkungen, Vergiftungen, stehen oft in einem ursächlichen Zusammenhang mit der Entstehung von Krebstumoren. Als besonders gefährlich gilt die Produktion von Gummi und Glas, Pestiziden und Kraftstoffen, Metallen und Textilien, Farben und Laborreagenzien. Gefährdet sind Arbeitnehmer in der Luft- und Raumfahrt, der Kohle- und Metallindustrie, in Chemie- und Nebenproduktfabriken, in der Baustoff- und Elektrodenindustrie, in der Kraftstoff- und Schmierstoffindustrie, in Kunststoff- und Monomerproduktionsanlagen.
- Luft-, Wasser- und Bodenverschmutzung sind für bis zu 4 % aller Krebserkrankungen weltweit verantwortlich. Karzinogene, die in großen Mengen in der Umwelt vorhanden sind, gelangen über die Atemluft, das Trinkwasser und die Nahrung in den Körper. Das Leben in ökologisch unsicheren Gebieten – in der Nähe großer Industrieanlagen und stark befahrener Verkehrsknotenpunkte – gilt als besonders gefährlich.
- Auch Infektionskrankheiten – insbesondere Virusinfektionen – können günstige Bedingungen für die Tumorentwicklung schaffen. Es ist wichtig, dies zu berücksichtigen und sich rechtzeitig impfen zu lassen sowie Infektions- und Parasitenerkrankungen vorzubeugen.
- Tabak- und Alkoholvergiftung gelten als Risikofaktoren für viele Krebsarten, nicht nur für Hirngliome.
- Unzureichende körperliche Aktivität, Übergewicht, falsche Ernährung, Stoffwechselstörungen, Kopfverletzungen, Gefäßerkrankungen – zusätzliche Stressfaktoren, die den Beginn intrazellulärer Störungen provozieren können.
- Im höheren Alter kommt es am häufigsten zu Neubildungen im Körper, daher sollten Menschen über 55 besonders auf ihre Gesundheit achten.
Der wichtigste und bedeutendste Risikofaktor für die Entwicklung eines Glioms bleibt jedoch die erbliche Veranlagung.
Pathogenese
Experten gehen derzeit von einer Reihe von Annahmen zur Entstehung von Hirngliomen aus. Jede Theorie hat ihre eigenen Grundlagen, doch den einzig richtigen und zuverlässigen pathogenetischen Mechanismus haben Wissenschaftler noch nicht identifiziert. In den meisten Fällen handelt es sich um folgende Faktoren bei der Entstehung von Neoplasien:
Embryogeneseversagen, das in der Störung der Organbildung und der Bildung „falscher“ Zellstrukturen besteht;
- Exposition gegenüber ionisierenden Strahlen, potenziellen Karzinogenen in Form von chemischen Stoffen, Lebensmittelzusatzstoffen usw.;
- Kopftrauma;
- Genetische Erkrankungen, die von Generation zu Generation weitergegeben werden („familiäres“ Gliom);
- Immunschwäche, Neuroinfektionen.
Die meisten Gliome wachsen diffus und dringen in das umgebende normale Hirngewebe ein. Je nach Malignitätsgrad kann sich der Tumor mehrere Jahre lang ohne Manifestation entwickeln. Bei einem aggressiven Verlauf verschlimmern sich die Symptome innerhalb weniger Monate rapide.
Ein Teil der Tumorentstehung ist auf dysembryogenetische Veränderungen zurückzuführen.
Der Hirnstamm kann auf verschiedenen Ebenen betroffen sein: Diffuse Hirnstammgliome unterscheiden sich sowohl anatomisch-morphologisch als auch klinisch. Einige dieser Neoplasien – insbesondere Gliome der Tetraplegieplatte – können relativ gutartig sein und keine Anzeichen einer Progression aufweisen. Ein Ponsgliom hingegen zeichnet sich durch seine besondere Bösartigkeit, Aggressivität und schlechte Prognose aus.
Diffuse Läsionen von Hirnstrukturen, bei denen mehr als drei anatomische Zonen der großen Hemisphären am pathologischen Prozess beteiligt sind, mit möglicher periventrikulärer Divergenz und Passage durch das Corpus, werden als Gliomatose bezeichnet. [ 4 ]
Ist ein Hirngliom erblich?
Ein gut belegtes Risiko für die Entstehung von Hirngliomen ist die Vererbung – das heißt das Vorhandensein ähnlicher oder anderer intrazerebraler Tumore bei direkten Vorfahren oder in der gleichen Generation. Radioaktive Belastungen und regelmäßiger oder längerer Kontakt mit potenziellen Karzinogenen verschlimmern die Situation.
Nicht nur Gliome können vererbt werden, sondern auch Krankheiten, die mit einem erhöhten Tumorwachstum ohne Bezug auf die Lokalisation einhergehen – insbesondere Neurofibromatose Typ 1 und 2, Li-Fraumeni-Syndrom, Hippel-Lindau-Syndrom. Oftmals finden sich in Gliomzellen Veränderungen in bestimmten Genen oder Chromosomen.
Die wichtigsten Pathologien, die mit der Entwicklung von Gliomen beim Menschen verbunden sind, sind in der Tabelle zusammengefasst:
Pathologie |
Chromosom |
Gen |
Vielzahl von Neoplasmen |
Li-Fraumeni-Syndrom |
17.13. |
TR53 |
Neuroektodermale Neoplasien, Astrozytom. |
Neurofibromatose |
17q11 |
NF1 |
Sehnervgliom, pilozytisches Astrozytom, Neurofibromatose |
Turcotte-Syndrom |
15:21 Uhr, 19:22 Uhr |
HMLH1, HPSM2 |
Astrozytom |
Tuberöse Sklerose (Burneville-Syndrom). |
9q34, 16p13 |
TSC1, TSC2 |
Gigantozelluläres subependymales Astrozytom |
Unabhängig von der Natur des Gliatumors, ob es sich um einen sporadischen Fall oder eine erbliche Pathologie handelt, handelt es sich um eine Erkrankung mit der Expression eines pathologisch veränderten Gens. Abgesehen von Neoplasien, die durch Lerneffekte entstehen, bleiben in anderen Situationen die Ursachen genetischer Veränderungen unklar.
Symptome Hirngliome
Die Merkmale der fokalen Symptomatologie hängen direkt vom Lokalisierungsbereich des Hirnglioms ab und sind eine Folge aller Arten von endokrinen Störungen, der Kompression des Nervengewebes oder lokaler destruktiver Prozesse.
Befindet sich das Neoplasma in der parietalen Zone, dominieren bei einer Person Erscheinungen wie Krampfanfälle, Sinnesstörungen und Hörbehinderungen.
Wenn das Gliom im Bereich der dominanten Hemisphäre lokalisiert ist, werden Sprachstörungen, Agraphie und Agnosie festgestellt.
Neoplasien des Temporallappens gehen häufig mit Krampfanfällen, Aphasie, Beeinträchtigung des Geruchs- und Sehsinns sowie Dyspnoe einher.
Bei erhöhtem Hirndruck kommt es zu einem entsprechenden Bild mit Gesichtsfeldeinschränkung, Augenmuskellähmung und Halbseitenlähmung.
Aufgrund der Spezifität des Tumorprozesses geht ein Hirngliom immer mehr oder weniger stark mit neurologischen Symptomen einher. Zunächst ist eine allgemeine Schwäche spürbar, der Patient möchte ständig schlafen, die Arbeitsfähigkeit ist beeinträchtigt, die Denkprozesse sind verlangsamt. In diesem Stadium besteht ein hohes Risiko, eine falsche Diagnose zu stellen und infolgedessen die falsche Behandlung zu verschreiben. Unter anderen unspezifischen Manifestationen:
- Vestibuläre Störungen, einschließlich unsicherem Gang, Gleichgewichtsverlust (z. B. beim Radfahren oder Treppensteigen), Taubheitsgefühl in den Gliedmaßen usw.;
- Allmähliche Verschlechterung des Sehvermögens, Verdoppelung des Sehbildes;
- Verschlechterung der Hörfunktion;
- Undeutliche Aussprache;
- Übelkeit und Erbrechen in Form von Anfällen unabhängig von Essen oder Trinken;
- Schwächung der mimischen Muskulatur und anderer Gesichtsmuskeln;
- Beschwerden beim Schlucken;
- Regelmäßige Kopfschmerzen (oft in den Morgenstunden).
Das Krankheitsbild weitet sich allmählich aus und verschlechtert sich: Bei manchen Patienten geschieht dies langsam, bei anderen abrupt, buchstäblich „vor ihren Augen“, innerhalb weniger Wochen. Im letzteren Fall handelt es sich um ein aggressives, sich schnell entwickelndes Gliom des Gehirns.
Erste Anzeichen
Gliome des Gehirns weisen in den frühen Entwicklungsstadien keine ausgeprägte Symptomatologie auf. Die ersten Manifestationen werden oft mit Anzeichen anderer, weniger gefährlicher Pathologien verwechselt.
Das klinische Bild eines Glioms ist im Allgemeinen vielfältig und wird durch die Lage und Größe des pathologischen Fokus bestimmt. Mit dem Wachstum des Neoplasmas entwickeln sich allgemeine zerebrale Symptome und nehmen zu:
- Anhaltende und regelmäßige Kopfschmerzen, die auf Standardmedikamente (nichtsteroidale Antirheumatika) nicht ansprechen;
- Zeitweise Übelkeit, manchmal bis zum Erbrechen;
- Ein unangenehmes, schweres Gefühl im Augapfelbereich;
- Krampfanfälle.
Zerebrale Manifestationen sind besonders intensiv, wenn der Tumor in die Ventrikel oder das Liquorsystem hineinwächst. Der Abfluss der Liquor cerebrospinalis ist beeinträchtigt, der intrakraniale Druck steigt an und es entwickelt sich ein Hydrozephalus. Der Prozess betrifft einen bestimmten Teil des Gehirns, was die Entwicklung der entsprechenden Klinik beeinflusst:
- Es gibt Probleme mit der Sehfunktion;
- Sprachbehinderung;
- Es treten Gleichgewichtsstörungen (Schwindel, gestörte Bewegungskoordination) auf;
- Paresen, Lähmungen der Arme, Beine;
- Gedächtnis und Konzentration sind beeinträchtigt;
- Denkprozesse sind beeinträchtigt;
- Es treten Verhaltensstörungen auf.
Im Anfangsstadium fehlen die Symptome praktisch oder sind so unbedeutend, dass sie keine Aufmerksamkeit erregen. Aus diesem Grund raten Experten dringend zu regelmäßigen Vorsorgeuntersuchungen und Kontrolluntersuchungen. Denn je früher der Tumorprozess erkannt wird, desto größer sind die Heilungs- und Überlebenschancen. [ 5 ]
Gliom des Gehirns bei einem Kind
Unter den vielen Hirntumoren im Kindesalter liegt der Anteil der Gliome zwischen 15 und 25 %. Kinder können im frühen Teenageralter und in den frühen Zwanzigern erkranken, Babys unter drei Jahren sind jedoch sehr selten betroffen.
Die Pathologie beginnt vor dem Hintergrund einer Mutation von Gliazellen. Bis heute gibt es keine Antwort auf die Frage, warum diese Mutation auftritt.
Als gesichert gilt lediglich, dass bestimmte Erbkrankheiten, die mit einem erhöhten Risiko für Tumorwachstum einhergehen, auch die Wahrscheinlichkeit erhöhen, an einem Hirngliom zu erkranken.
Darüber hinaus haben Wissenschaftler herausgefunden, dass Gliazellen Abweichungen in einzelnen Genen oder Chromosomen aufweisen können. Aufgrund dieser Störung setzt ein Mutationsmechanismus ein, der nicht erblich ist. Es ist möglich, dass dies in einem der frühesten Entwicklungsstadien geschieht.
Es ist erwiesen, dass das Vorliegen einer akuten Leukämie oder eines Retinoblastoms in der Krankengeschichte des Kindes oder eine Bestrahlung des Gehirns aus anderen Gründen das Risiko der Gliombildung (nach einer gewissen Zeit) deutlich erhöht.
Die Symptomatik im Kindesalter hängt vom Malignitätsgrad und der Lokalisation des pathologischen Fokus ab. Man unterscheidet zwischen spezifischen und unspezifischen Symptomen:
- Unspezifische Symptome sind nicht an den Bereich gebunden, in dem sich das Gliom befindet. Häufige Symptome sind Kopfschmerzen, Schwindel, Appetitlosigkeit, Erbrechen ohne Zusammenhang mit der Nahrungsaufnahme, Gewichtsverlust (aus unbekannten Gründen), ständige Müdigkeit, Leistungsabfall, Konzentrationsschwierigkeiten und Verhaltensstörungen. Diese Symptome sind auf eine Kompression intrakranieller Strukturen zurückzuführen, die durch den direkten Druck der wachsenden Masse und eine Störung der Zerebrospinalflüssigkeitszirkulation erklärt werden kann. Es besteht das Risiko einer zerebralen Hydrozele.
- Die spezifische Symptomatologie hängt von der unmittelbaren Lokalisation des glialen pathologischen Fokus ab. Beispielsweise geht ein Kleinhirntumor bei Kindern meist mit Gang- und Gleichgewichtsstörungen einher. Die Läsion des Großhirns äußert sich in Krampfanfällen und das Tumorwachstum im Rückenmark in einer Lähmung der Muskulatur. Es kommt vor, dass sich das Sehvermögen des Babys stark verschlechtert, das Bewusstsein gestört ist, der Schlaf beeinträchtigt ist oder andere Entwicklungsprobleme auftreten.
In der Regel zeigt sich ein bösartiges Gliom im Kindesalter innerhalb weniger Wochen oder Monate seiner Entwicklung: Oft ist es durch ein schnelles und unkontrolliertes Wachstum des Neoplasmas gekennzeichnet.
Kinder mit bösartigen Gliatomen werden von Ärzten in pädiatrischen Klinikzentren behandelt, die auf pädiatrische Onkologie spezialisiert sind. In der Regel kommen chirurgische Behandlungen, Bestrahlung und Chemotherapie zum Einsatz.
Der wichtigste Behandlungsschritt ist die Neurochirurgie. Je radikaler sie durchgeführt wird, desto besser sind die Heilungschancen des Kindes. Ein chirurgischer Eingriff ist jedoch nicht immer möglich: Insbesondere bei der Entfernung von Hirnstammgliomen sowie bei der Bestrahlung von Kindern unter drei Jahren können Probleme auftreten.
Gliome des Zentralhirns (Zwischen- und Mittelhirn) lassen sich nur schwer vollständig entfernen, da die Gefahr einer Schädigung gesunden Gewebes besteht. Ist eine vollständige Resektion des Tumors nicht möglich, wird dem Patienten eine palliative Behandlung verordnet.
Kinder mit bösartigen Gliomen werden nach standardisierten Protokollen behandelt, die in streng kontrollierten klinischen Studien ermittelt wurden. Die gängigsten Protokolle sind:
- HIT HGG 2007: umfasst die Behandlung von Kindern im Alter von 3-17 Jahren.
- HIT SKK: Geeignet für Kleinkinder (bis drei Jahre) und ohne Strahlenbehandlung.
Die Überlebensstatistiken für Gliome bei Kindern sind im Allgemeinen nicht sehr optimistisch. Die Wirksamkeit der Behandlungsmaßnahmen für ein bestimmtes Kind lässt sich jedoch in keinem Fall im Voraus vorhersagen. Es ist wichtig, alle Anweisungen des Arztes sorgfältig zu befolgen, da dies die Heilungschancen deutlich erhöht.
Formen
Gliome können niedrigmaligne und hochmaligne sein, mit intensivem Wachstum und einer Neigung zur Metastasierung. Es ist wichtig zu verstehen, dass niedrige Malignität nicht gleichbedeutend mit Tumorsicherheit ist. Jede Hirnneoplasie erzeugt zusätzliches Volumen, quetscht Hirnstrukturen, was zu deren Verschiebung und erhöhtem Hirndruck führt. Infolgedessen kann der Patient sterben.
Es gibt zwei Haupttypen maligner Astrozytome: Glioblastome und anaplastische Astrozytome, die nach molekularen Veränderungen unterteilt werden. Sekundäre maligne Tumoren, die sich aus Astrozytomen entwickeln und einen geringen Malignitätsgrad aufweisen, treten am häufigsten bei jungen Patienten auf. Initial maligne Gliatumoren treten häufiger bei älteren Patienten auf.
Je nach struktureller Lokalisation unterscheidet man zwischen:
- Supratentorielle (mit Lokalisierung oberhalb des Kleinhirns im Bereich der Seitenventrikel, der großen Hemisphären);
- Subtentoriell (mit Lokalisation unterhalb des Kleinhirns in der hinteren Schädelgrube).
Nach histologischen Merkmalen werden folgende Arten von Gliomen unterschieden:
- Das astrozytische Gliom ist das häufigste. Es wird wiederum in noduläre und diffuse Gliome unterteilt (letztere zeichnen sich durch schnelles Wachstum und ein Schlaganfallmuster aus).
- Oligodendrogliom – tritt bei 5 % der Patienten auf. Es weist Verkalkungen auf, meist im Frontallappen.
- Ependymgliom – wächst aus den Strukturen, die die Wände des Zentralkanals des Rückenmarks und der Ventrikel auskleiden. Wächst oft in die Dicke der Hirnsubstanz sowie in das Lumen des Gehirns hinein.
Auch gemischte pathologische Herde wie Subependymom, Oligoastrozytom etc. sind möglich.
Alle Gliome werden in die folgenden Stadien eingeteilt:
- Langsam wachsende, relativ gutartige Neubildungen ohne offensichtliche klinische Symptome.
- Langsam wachsende „Borderline“-Gliome, die sich allmählich in Stadium III und höher verwandeln.
- Bösartiges Gliom.
- Bösartiges Gliom mit intensivem, aggressivem Wachstum und Ausbreitung und schlechter Prognose.
Je niedriger das Malignitätsstadium, desto geringer ist die Wahrscheinlichkeit einer Metastasierung und eines Wiederauftretens des entfernten Neoplasmas und desto größer sind die Heilungschancen des Patienten. Die größte Gefahr stellt das Glioblastoma multiforme dar, ein niedrig differenzierter Prozess mit intensivem Wachstum und Entwicklung. [ 6 ]
Mögliche und häufigste Varianten des Neuroglioms:
- Gliome mit Hirnstamm- und Brückenläsionen befinden sich im Übergang vom Gehirn zum Rückenmark. Dort befinden sich wichtige Neurozentren, die für die Atmung, das Herz und die Motorik verantwortlich sind. Ist dieser Bereich beschädigt, ist die Funktion des Vestibular- und Sprachapparates gestört. Häufig wird es bereits im Kindesalter diagnostiziert.
- Visuelles Gliom betrifft Neurogliazellen, die den Sehnerv umgeben. Die Erkrankung verursacht Sehstörungen und Exophthalmus. Sie tritt häufiger bei Kindern auf.
- Das niedrig maligne Neurogliom ist durch langsames Wachstum gekennzeichnet und befindet sich häufiger in den großen Hemisphären und im Kleinhirn. Es tritt häufiger bei jungen Menschen (Jugendlichen und jungen Erwachsenen um das 20. Lebensjahr) auf.
- Gliome des Corpus callosum treten häufiger bei Personen zwischen 40 und 60 Jahren auf und werden am häufigsten durch ein Glioblastom repräsentiert.
- Das Chiasmagliom ist in der Sehnervenübergangszone lokalisiert und geht daher mit Myopie, Gesichtsfeldausfall, okklusivem Hydrozephalus und neuroendokrinen Störungen einher. Es kann in jedem Alter auftreten, betrifft aber am häufigsten Patienten mit Neurofibromatose Typ I.
Komplikationen und Konsequenzen
Gliome mit geringer Malignität (Grad I–II, hochmaligne – z. B. Astrozytom, Oligoastrozytom, Oligodendrogliom, pleomorphes Xanthoastrozytom usw.) und hoher Malignität (Grad III–IV – Glioblastom, anaplastisches Oligodendrogliom, Oligoastrozytom und Astrozytom). Gliome mit Grad IV sind besonders bösartig.
Das Hirnstammgliom hat eine sehr ungünstige Prognose, gerade weil der Tumor eine Hirnregion befällt, in der die wichtigsten Nervenverbindungen zwischen Gehirn und Gliedmaßen konzentriert sind. Schon ein relativ kleiner Tumor in diesem Bereich kann den Zustand des Patienten schnell verschlechtern und Lähmungen hervorrufen.
Nicht weniger ungünstige Folgen treten auf, wenn andere Hirnregionen betroffen sind. Oft handelt es sich um einen Tumor der Großhirnrinde, der dem Patienten trotz Behandlung keine Chance auf ein langes Leben lässt. Oftmals kann nur der Tod hinausgezögert werden.
Laut medizinischer Statistik beträgt die Fünfjahresüberlebensrate oft nur 10–20 %. Diese Zahlen hängen jedoch maßgeblich vom Grad der Malignität sowie der genauen Lokalisation und dem Umfang des durchgeführten chirurgischen Eingriffs ab. Nach vollständiger Entfernung des pathologischen Fokus erhöht sich die Überlebensrate signifikant (manchmal bis zu 50 %). Eine fehlende oder unmögliche Behandlung führt garantiert zum Tod des Patienten.
Die Mehrzahl der niedrigmalignen Gliomatumore kann in das Hirngewebe eindringen und sich im Laufe mehrerer Jahre bösartig entwickeln.
Das Risiko eines Gliomrückfalls wird von Experten als „sehr wahrscheinlich“ eingeschätzt. Dennoch sollte die Behandlung nicht vernachlässigt werden: Es ist wichtig, so lange wie möglich eine gute Lebensqualität zu gewährleisten.
Rezidivierende Gliome haben grundsätzlich eine schlechtere Prognose als Primärtumoren. Moderne Behandlungsprotokolle, die auf Therapieoptimierungsstudien basieren, erzielen jedoch selbst bei hochmalignen Neoplasien oft ausreichend gute Ergebnisse für Patienten.
Mögliche Ergebnisse nach einer Chemotherapie:
- Abmagerung, Abmagerung, Verdauungsstörungen, Erkrankungen der Mundhöhle;
- Erhöhte Erregbarkeit des zentralen Nervensystems, Asthenie;
- Verschlechterung der Hörfunktion, Tinnitus und Ohrensausen;
- Krampfanfälle, depressive Störungen;
- Hypertensive Krise, Veränderung des Blutbildes;
- Nierenversagen;
- Allergische Prozesse, Haarausfall, Auftreten von Pigmentflecken am Körper.
Nach einer Chemotherapie stellen die Patienten eine deutliche Schwächung des Immunsystems fest, die zur Entwicklung verschiedener Infektionskrankheiten führen kann.
Diagnose Hirngliome
Folgende Anzeichen weisen auf ein Hirngliom hin:
- Der Patient leidet unter lokalisierten oder generalisierten Anfällen, die für die kortikale Lage des Neoplasmas und seine langsame Entwicklung charakteristisch sind. Epi-Anfälle treten bei 80 % der Patienten mit niedriggradigen Gliomen und bei 30 % der Patienten mit hochgradigen Gliomen auf.
- Erhöhter intrakranieller Druck ist besonders charakteristisch für Tumoren im rechten Frontal- und Parietallappen. Die damit verbundene Störung der Blutzirkulation und des Liquorkreislaufs führt zu ständigen und zunehmenden Kopfschmerzen, Übelkeit mit Erbrechen, Sehstörungen und Schläfrigkeit. Es kommt zu einem Ödem des Sehnervs und einer Lähmung des ableitenden Nervs. Ein Anstieg des intrakraniellen Drucks auf kritische Werte kann zu Koma und Tod führen. Eine weitere Ursache für einen hohen Augeninnendruck ist ein Hydrozephalus.
- Der Patient hat ein wachsendes Herdbild. In supratentoriellen Formationen sind die motorischen und sensorischen Bereiche gestört, Hemiopie, Aphasie und kognitive Störungen schreiten voran.
Bei Verdacht auf ein Hirntumor ist eine MRT mit oder ohne Kontrastmittel (Gadolinium) optimal, um dessen Lage, Größe und weitere Merkmale zu bestimmen. Ist eine Magnetresonanztomographie nicht möglich, wird eine Computertomographie durchgeführt, zur Differenzierung dient die Magnetresonanzspektroskopie. Trotz der Aussagekraft dieser diagnostischen Methoden wird die endgültige Diagnose erst nach histologischer Bestätigung im Rahmen der Resektion des Tumorherdes gestellt.
Angesichts der oben genannten Kriterien empfiehlt es sich, die Diagnose mit einer gründlichen Anamnese, einer Beurteilung des somato-neurologischen Status und des funktionellen Status zu beginnen. Der neurologische Status wird zusammen mit der Feststellung möglicher intellektueller und mnestischer Störungen beurteilt.
Empfohlene Labortests:
- Eine umfassende allgemeine klinische Blutuntersuchung;
- Eine vollständige Blutchemieuntersuchung;
- Urinanalyse;
- Blutgerinnungsstudie;
- Analyse auf onkologische Marker (AFP, Beta-hCG, LDH – relevant bei Verdacht auf eine Läsion der Zirbeldrüsenzone).
Zur Abklärung der Prognose bei Patienten mit Glioblastom und anaplastischem Astrozytom werden die IDH1|2-1-Genmutation und die MGMT-Genmethylierung untersucht. Bei Patienten mit Oligodendrogliom und Oligoastrozytom wird die 1p|19q-Kodierung bestimmt.
Die instrumentelle Diagnostik wird in erster Linie durch die obligatorische Magnetresonanztomographie des Gehirns (manchmal auch des Rückenmarks) dargestellt. Die MRT wird in drei Projektionen im Standardmodus T1-2, FLAIR, T1 mit Kontrast durchgeführt.
Bei entsprechender Indikation werden Ultraschalluntersuchungen des Gefäßnetzes, funktionelle Magnetresonanztomographien des Motor- und Sprachbereichs sowie Angiographien, Spektroskopie, MR-Traktographie und Perfusion durchgeführt.
Zusätzliche Untersuchungen können Folgendes umfassen:
- Elektroenzephalographie des Gehirns;
- Konsultationen mit einem Neurochirurgen, Onkologen, Radiologen, Augenarzt, Radiologen.
Differenzialdiagnose
Eine Differentialdiagnose muss unbedingt bei nicht-tumorösen Pathologien durchgeführt werden – insbesondere bei Blutungen, die durch arteriovenöse oder arterielle Fehlbildungen verursacht werden, sowie bei pseudotumorösen demyelinisierenden Prozessen, entzündlichen Erkrankungen (Toxoplasmose, Hirnabszess usw.).
Darüber hinaus ist zwischen dem primären Tumorherd und Metastasen im zentralen Nervensystem zu unterscheiden.
Mit modernen Möglichkeiten der Magnetresonanztomographie ist es möglich, diagnostische Maßnahmen ausreichend genau durchzuführen, um den Ursprung des primären Fokus im ZNS herauszufinden. Die MRT des Gehirns wird mit oder ohne Kontrastmittel im T1-, T2-FLAIR-Modus – in drei Projektionen – oder in dünnen Schichten in axialer Projektion (SPGR-Modus) durchgeführt. Diese Diagnosemethoden ermöglichen die genaue Bestimmung von Lage, Größe, strukturellen Merkmalen des Neoplasmas sowie seiner Beziehung zum Gefäßnetz und benachbarten Hirnarealen.
Zusätzlich können zur Differentialdiagnose eine CT (mit oder ohne Kontrastmittel), eine CT-Angiographie (MR-Angiographie), eine MR-Traktographie, eine MR- oder CT-Perfusion durchgeführt werden. Bei entsprechender Indikation wird eine CT/PET des Gehirns mit Methionin, Cholin, Tyrosin und anderen Aminosäuren eingesetzt.
Behandlung Hirngliome
Die spezifische Therapie besteht aus chirurgischen, chemotherapeutischen und radiologisch-radiologischen Maßnahmen. Obligatorisch ist, wenn möglich, eine vollständige Resektion des Tumorherdes, die eine schnelle Linderung der Symptome und eine histologische Sicherung der Diagnose ermöglicht.
Die Bestrahlung erhöht die Lebenserwartung der Patienten. Standardmäßig wird eine Gesamtdosis von 58 bis 60 Gy verabreicht, aufgeteilt auf Einzeldosen von 1,8–2 Gy. Der Tumor wird lokal bestrahlt, wobei zusätzlich ein Umkreis von bis zu 3 cm erfasst wird. Die Strahlentherapie ist im Vergleich zur Brachytherapie verträglicher. In manchen Fällen werden radiochirurgische Methoden empfohlen, die aus der Bestrahlung mit einem Gamma Knife oder einem linearen Gaspedal sowie einer Neutroneneinfang-Bor-Therapie bestehen.
Die Notwendigkeit einer adjuvanten Chemotherapie ist umstritten. In einigen Fällen konnten Nitrosoharnstoffpräparate die Lebenserwartung der Patienten um bis zu eineinhalb Jahre verlängern, einige Ergebnisse der Anwendung solcher Chemopräparate waren jedoch negativ. Heute werden aktiv Zytostatika, neoadjuvante Therapie (vor der Bestrahlung), Kombinationspräparate, intraarterielle Chemotherapie oder Hochdosis-Chemotherapie mit anschließender Stammzelltransplantation eingesetzt.
Generell ist für eine erfolgreiche Behandlung von Gliomen ein umfassender Ansatz sehr wichtig, dessen Umfang von der Lokalisation und dem Grad der Bösartigkeit der Masse, ihrer Größe und dem allgemeinen Gesundheitszustand des Patienten abhängt.
Bei Hirnstammgliomen werden chirurgische Eingriffe selten durchgeführt. Die Hauptkontraindikation für eine Operation ist die Lokalisation des Herdes – in unmittelbarer Nähe zu lebenswichtigen Teilen. In einigen Fällen ist es möglich, ein Gliom des Rumpfes mikrochirurgisch mit prä- und postoperativer Chemotherapie zu entfernen. Ein solcher Eingriff ist sehr komplex und erfordert spezielle Qualifikationen eines Neurochirurgen.
Strahlenchirurgie und insbesondere stereotaktische Chirurgie mit hohen ionisierenden Dosen sind sehr effektiv. Der Einsatz dieser Technik in frühen Stadien der Neoplasmaentwicklung ermöglicht manchmal eine verlängerte Remission oder sogar eine vollständige Heilung des Patienten.
Strahlentherapie wird häufig mit Chemotherapie kombiniert, was die Wirksamkeit der Interventionen verbessert und die Strahlenbelastung reduziert. Bei Gliomen sind nicht alle chemopräventiven Mittel therapeutisch erfolgreich, daher werden sie einzeln verordnet und die Dosierung gegebenenfalls angepasst.
Um Schmerzen zu lindern und den Hirndruck zu senken, wird unabhängig von der Hauptbehandlung eine symptomatische Therapie verordnet – insbesondere Kortikosteroide, Analgetika und Beruhigungsmittel.
Medikamente
Kortikosteroide wirken auf Schwellungen und reduzieren die Schwere der neurologischen Symptome für mehrere Tage. Aufgrund vielfältiger Nebenwirkungen und einer erhöhten Wahrscheinlichkeit unerwünschter Wechselwirkungen mit Chemotherapeutika werden jedoch nur minimal wirksame Dosen von Steroiden verwendet und diese so schnell wie möglich (z. B. nach einer Operation) abgesetzt.
Antikonvulsiva werden systematisch als sekundäre Präventionsmaßnahme bei Patienten eingesetzt, die bereits epileptische Anfälle erlitten haben. Diese Medikamente können schwerwiegende Nebenwirkungen verursachen und zudem Wechselwirkungen mit Chemotherapeutika hervorrufen.
Antikoagulanzien sind insbesondere im postoperativen Stadium relevant, da das Risiko einer Thrombophlebitis bei Gliomen recht hoch ist (bis zu 25 %).
Von der Einnahme von Antidepressiva und Anxiolytika wird eine gute Wirkung erwartet. Die Einnahme von Methylphenidat 10–30 mg/Tag in zwei Dosen ermöglicht häufig die Optimierung der kognitiven Fähigkeiten, die Verbesserung der Lebensqualität und die Aufrechterhaltung der Arbeitsfähigkeit.
Neurologische Ausfälle und Anzeichen eines Hirnödems (Kopfschmerzen, Bewusstseinsstörungen) werden durch Kortikosteroide – insbesondere Prednisolon oder Dexamethason – beseitigt. |
Das Schema und die Dosierung von Kortikosteroiden werden individuell ausgewählt, wobei die minimal wirksame Dosis eingehalten wird. Am Ende des Behandlungsverlaufs werden die Medikamente schrittweise abgesetzt. |
Kortikosteroide werden zusammen mit magenschützenden Medikamenten eingenommen – Protonenpumpenblockern oder H2-Histaminblockern. |
Diuretika (Furosemid, Mannitol) werden bei starken Schwellungen und Verschiebungen der Gehirnstrukturen als Ergänzung zu Kortikosteroiden verschrieben. |
Bei Krampfanfällen (einschließlich Anamnese) oder epileptiformen Symptomen im Elektroenzephalogramm wird zusätzlich eine antikonvulsive Therapie verordnet. Antikonvulsiva werden nicht zur Prophylaxe verschrieben. |
Patienten mit Indikationen für eine Chemotherapie wird die Einnahme von Antikonvulsiva empfohlen, die die Leberenzymfunktion nicht beeinträchtigen. Medikamente der Wahl: Lamotrigin, Valproinsäure, Levetiracetam. Nicht anzuwenden sind: Carbamazepin, Phenobarbital. |
Kopfschmerzen bei Hirngliomen werden mit einer Kortikosteroidbehandlung behandelt. |
In einigen Fällen von Kopfschmerzen können nichtsteroidale Antirheumatika oder Tramadol eingesetzt werden. |
Wenn der Patient nichtsteroidale entzündungshemmende Medikamente einnimmt, werden diese einige Tage vor der Operation abgesetzt, um das Risiko von Blutungen während der Operation zu minimieren. |
In bestimmten Schmerzfällen können narkotische Analgetika – wie Fentanyl oder Trimeperidin – empfohlen werden. |
Zur Vorbeugung einer Lungenembolie ab dem dritten postoperativen Tag wird die Gabe niedermolekularer Heparine – insbesondere Enoxaparin-Natrium oder Nadroparin-Calcium – verordnet. |
Wenn der Patient eine systematische Behandlung mit Antikoagulanzien oder Thrombozytenaggregationshemmern erhält, wird er spätestens eine Woche vor dem chirurgischen Eingriff auf niedermolekulare Heparine umgestellt. Einen Tag vor der Operation wird die Behandlung abgesetzt und 24–48 Stunden nach der Operation wieder aufgenommen. |
Wenn ein Patient mit Gliom eine Venenthrombose der unteren Extremitäten aufweist, wird eine Behandlung mit direkten Antikoagulanzien durchgeführt. Die Möglichkeit der Platzierung eines CAVA-Filters ist nicht ausgeschlossen. |
Chemotherapie bei bösartigen Gliomen des Gehirns
Als grundlegende Antitumor-Chemotherapieschemata für Gliome gelten:
- Lomustin 100 mg/m² am ersten Tag, Vincristin 1,5 mg/m² am ersten und achten Tag, Procarbazin 70 mg/m² vom achten bis zum einundzwanzigsten Tag, Behandlungen alle sechs Wochen.
- Lomustin 110 mg/m² alle sechs Wochen.
- Temozolomid 5/23 150 bis 200 mg/m² vom ersten bis zum fünften Tag, alle 28 Tage.
- Temozolomid als Teil einer Radiochemotherapie, 75 mg/m² an jedem Strahlentag.
- Temozolomid mit Cisplatin oder Carboplatin (80 mg/m²) und Temozolomid 150–200 mg/m² an den Tagen 1 bis 5 alle 4 Wochen.
- Temozolomid 7/7 mit 100 mg/m² an den Tagen 1–8 und 15–22 der Behandlung, mit Wiederholung alle vier Wochen.
- Bevacizumab 5 bis 10 mg/kg an Tag eins und fünfzehn und Irinotecan 200 mg/m² an Tag eins und fünfzehn, alle vier Wochen wiederholt.
- Bevacizumab 5 bis 10 mg/kg an den Tagen eins, fünfzehn und neunundzwanzig und Lomustin 90 mg/m² am ersten Tag alle sechs Wochen.
- Bevacizumab 5 bis 10 mg/kg an den Tagen eins und fünfzehn, Lomustin 40 mg an den Tagen eins, acht, fünfzehn und zweiundzwanzig, Wiederholung alle sechs Wochen.
- Bevacizumab 5 bis 10 mg/kg an Tag eins und fünfzehn, alle vier Wochen wiederholt.
Zytostatika hemmen in vielen Fällen erfolgreich das Wachstum von Tumorzellen, zeigen jedoch keine Selektivität gegenüber gesunden Geweben und Organen. Daher haben Experten eine Reihe von Kontraindikationen identifiziert, bei denen eine Chemotherapie des Glioms nicht möglich ist:
- Übermäßige individuelle Empfindlichkeit gegenüber chemopräventiven Mitteln;
- Dekompensation der Herz-, Nieren- und Leberfunktion;
- Verminderte Hämatopoese im Knochenmark;
- Probleme mit der Nebennierenfunktion.
Die Chemotherapie wird mit äußerster Vorsicht verabreicht:
- Patienten mit erheblichen Herzrhythmusstörungen;
- Bei Diabetes;
- Bei akuten Virusinfektionen;
- An ältere Patienten;
- Patienten, die an chronischem Alkoholismus (chronischer Alkoholintoxikation) leiden.
Die schwerwiegendste Nebenwirkung chemopräventiver Medikamente ist ihre Toxizität: Zytostatika beeinträchtigen gezielt die Funktionsfähigkeit von Blutzellen und verändern deren Zusammensetzung. Die Folge ist eine Abnahme der Thrombozyten- und Erythrozytenmasse und die Entstehung einer Anämie.
Bevor ein Arzt einem Patienten eine Chemotherapie verschreibt, berücksichtigt er stets den Grad der Toxizität der Medikamente und die wahrscheinlichen Komplikationen nach ihrer Anwendung. Chemotherapie-Kurse werden stets sorgfältig von Spezialisten und regelmäßigen Blutuntersuchungen überwacht.
Mögliche Folgen einer Zytostatikatherapie:
- Magerkeit, Abmagerung;
- Schluckbeschwerden, trockene Schleimhäute, Parodontitis, Dyspepsie;
- Instabilität des zentralen Nervensystems, manisch-depressive Störungen, Krampfanfallssyndrom, Asthenie;
- Verschlechterung der Hörfunktion;
- Anstieg des Blutdrucks bis hin zur Entwicklung einer hypertensiven Krise;
- Abnahme der Blutplättchen, der roten Blutkörperchen, der weißen Blutkörperchen, multiple Blutungen, innere und äußere Blutungen;
- Nierenversagen;
- Allergische Prozesse;
- Haarausfall, Auftreten von Bereichen mit erhöhter Pigmentierung.
Nach Chemotherapie-Zyklen besteht bei den Patienten ein erhöhtes Risiko, an Infektionskrankheiten zu erkranken. Muskel- und Gelenkschmerzen treten häufig auf.
Um das Risiko unerwünschter postchemotherapeutischer Wirkungen zu verringern, sind weitere Rehabilitationsmaßnahmen erforderlich, deren Ziel die Wiederherstellung eines normalen Blutbildes, die Stabilisierung der Herz-Kreislauf-Aktivität und die Normalisierung des neurologischen Status ist. Ausreichende psychologische Unterstützung ist unbedingt erforderlich.
Chirurgische Behandlung
Ziel der Operation ist es, den Tumorherd möglichst weit zu entfernen, wodurch der intrakraniale Druck gesenkt, die neurologische Insuffizienz verringert und das notwendige Biomaterial für die Forschung bereitgestellt werden soll.
- Die Operation wird in einer spezialisierten neurochirurgischen Abteilung oder Klinik durchgeführt, deren Spezialisten über Erfahrung in neuroonkologischen Eingriffen verfügen.
- Der Chirurg legt den Zugang mittels plastischer Knochentrepanation im Bereich der vermuteten Gliomlokalisation an.
- Liegt das Neoplasma anatomisch in der Nähe von motorischen Bereichen oder Bahnen, in den Kernen oder entlang von Hirnnerven, wird eine intraoperative neurophysiologische Überwachung durchgeführt.
- Neuronavigationssysteme, intraoperative Fluoreszenznavigation mit 5-Aminolävulinsäure ist wünschenswert, um die Entfernung des Neoplasmas zu maximieren.
- Nach dem Eingriff wird am 1.-2. Tag eine Kontroll-CT oder MRT (mit oder ohne Kontrastmittelgabe) durchgeführt.
Ist eine chirurgische Resektion eines Glioms nicht möglich oder zunächst als nicht sinnvoll erkannt oder besteht der Verdacht auf ein Lymphom des Zentralnervensystems, wird eine Biopsie (offen, stereotaktisch, mit Navigationsüberwachung etc.) durchgeführt. |
Bei Patienten mit zerebraler Gliomatose erfolgt die Diagnose durch eine stereotaktische Biopsie, da die therapeutische Vorgehensweise weitgehend vom histologischen Bild abhängt. |
In bestimmten Situationen – bei älteren Patienten, bei schweren neurologischen Erkrankungen, bei der Lokalisation des Glioms im Rumpf und anderen lebenswichtigen Teilen – wird die Behandlung nach einer allgemeinen ärztlichen Konsultation auf der Grundlage von Symptomen und bildgebenden Informationen geplant. |
Bei Patienten mit piloidem Astrozytom sowie nodulären Formen von Hirnstammneoplasien und exophytischen Prozessen wird eine Resektion oder offene Biopsie empfohlen. |
Patienten mit diffusem Ponsgliom und anderen diffusen Neoplasien des Rumpfes werden mit Strahlentherapie und antitumoraler medikamentöser Therapie behandelt. Eine Verifizierung ist in diesen Fällen nicht erforderlich. |
Patienten mit einem tetraplegischen Plattengliom werden nach der Entfernung der zerebralen Hydrozele einer systematischen Magnetresonanztomographie und klinischen Überwachung unterzogen. Wenn das Neoplasma Anzeichen von Wachstum zeigt, wird es durch weitere Bestrahlung entfernt. |
Wenn eine Teilresektion oder Biopsie eines niedrigmalignen Glioms durchgeführt wird, werden Patienten mit zwei oder mehr Risikofaktoren zwangsläufig mit Strahlen- und/oder Chemotherapie behandelt. |
Bei Patienten mit subependymalem Riesenzellastrozytom ist eine vollständige Resektion zwingend erforderlich. |
Everolimus wird bei diffusem subependymalem Riesenzellastrozytom verschrieben. |
Um die Qualität der radikalen Resektion des Tumorgewebes zu klären, sollte nach dem Eingriff ein piloides Astrozytom mittels Magnetresonanztomographie entfernt werden. |
Beim Glioblastom sollte die postoperative Therapie (Bestrahlung + Chemotherapie) mit der Gabe von Temozolomid kombiniert werden. |
Bei einem anaplastischen Astrozytom ist nach der Operation eine Strahlentherapie mit weiterer medikamentöser Therapie angezeigt. Zum Einsatz kommen Lomustin, Temozolomid. |
Patienten mit anaplastischem Oligodendrogliom oder Oligoastrozytom erhalten nach der Operation sowohl eine Strahlen- als auch eine Chemotherapie (Temozolomid- oder PCV-Monotherapie). |
Bei älteren Patienten mit ausgedehntem hochmalignen Gliom erfolgt die Bestrahlung im hypofraktionierten Modus oder es wird eine Monotherapie mit Temozolomid durchgeführt. |
Im Falle eines Gliomrezidivs werden die Möglichkeit einer erneuten Operation und die anschließende Behandlungstaktik von einem Facharztkonsilium besprochen. Das optimale Behandlungsschema bei Rezidiven: erneute Operation + systemische Chemotherapie + wiederholte Strahlenexposition + palliative Maßnahmen. Bei lokalisierten kleineren Bereichen mit wiederkehrendem Tumorwachstum kann eine Radiochirurgie eingesetzt werden. |
Die Medikamente der Wahl bei wiederkehrendem Gliomwachstum sind Temozolomid und Bevacizumab. |
Das Wiederauftreten hochmaligner Oligodendrogliome und anaplastischer Astrozytome ist eine Indikation für eine Behandlung mit Temozolomid. |
Das pleomorphe Xanthoastrozytom wird ohne obligatorische adjuvante Chemotherapie entfernt. |
Eine Besonderheit von Gliomen ist die Schwierigkeit ihrer Behandlung und Entfernung. Der Chirurg versucht, das Gewebe des Neoplasmas möglichst vollständig zu entfernen, um den Zustand zu kompensieren. Viele Patienten können ihre Lebensqualität verbessern und verlängern, doch bei hochmalignen Tumoren bleibt die Prognose ungünstig: Es besteht eine erhöhte Wahrscheinlichkeit eines erneuten Wachstums des pathologischen Fokus.
Ernährung bei Gliomen des Gehirns
Die Ernährung von Patienten mit bösartigen Tumoren ist ein wichtiger Punkt, dem leider viele Menschen nicht viel Aufmerksamkeit schenken. Dank einer Ernährungsumstellung ist es jedoch möglich, die Entwicklung von Gliomen zu verlangsamen und das geschwächte Immunsystem zu stärken.
Wichtige Bereiche der Ernährungsumstellung:
- Normalisierung von Stoffwechselprozessen, Stärkung der Immunabwehr;
- Entgiftung des Körpers;
- Optimierung des Energiepotenzials;
- Sicherstellung der normalen Funktion aller Organe und Systeme des Körpers während einer für sie so schwierigen Zeit.
Eine rationale und ausgewogene Ernährung ist sowohl für Patienten mit frühen Stadien niedrig maligner Neoplasien als auch für Patienten mit dem letzten Stadium eines Glioblastoms erforderlich. Eine sorgfältig ausgewählte Ernährung trägt zur Verbesserung des allgemeinen Wohlbefindens und zur Wiederherstellung geschädigter Gewebe bei, was insbesondere vor dem Hintergrund der Zytostatika- und Strahlentherapie wichtig ist. Das Gleichgewicht der Nährstoffkomponenten und die richtigen Stoffwechselprozesse verhindern die Bildung von Infektionsherden, blockieren Entzündungsreaktionen und beugen der Erschöpfung des Körpers vor.
Bei einem Hirngliom werden folgende Nahrungsmittel und Getränke empfohlen:
- Rot, gelb und orange gefärbtes Obst und Gemüse (Tomaten, Pfirsiche, Aprikosen, Karotten, Rüben, Zitrusfrüchte) mit Carotinoiden, die gesunde Zellen vor den negativen Auswirkungen der Strahlentherapie schützen;
- Kohl (Blumenkohl, Brokkoli, Rosenkohl), Radieschen, Senf und andere Pflanzenprodukte, die Indol enthalten – einen Wirkstoff, der schädliche toxische und chemische Faktoren neutralisiert;
- Grünes (Dill, Petersilie, junge Löwenzahn- und Brennnesselblätter, Rhabarber, Rucola, Spinat), grüne Erbsen und Spargel, Spargelbohnen und Algen (Seetang, Spirulina, Chlorella);
- Grüner Tee;
- Knoblauch, Zwiebeln, Ananas, die eine Antitumor- und Entgiftungswirkung haben;
- Kleie, Getreide, Vollkornbrot, gekeimte Sprossen von Hülsenfrüchten, Getreide und Samen;
- Dunkle Trauben, Himbeeren, Erdbeeren und Erdbeeren, Heidelbeeren, Brombeeren, Granatäpfel, Johannisbeeren, schwarze Johannisbeeren, Vogelbeeren, Heidelbeeren, Sanddorn, Kirschen und andere Beeren, die natürliche Antioxidantien enthalten, die die negativen Auswirkungen von freien Radikalen, Viren und Karzinogenen reduzieren;
- Fettarme Milchprodukte.
Sie sollten das Verdauungssystem und den gesamten Körper nicht mit schweren und fetthaltigen Lebensmitteln belasten. Es ist sinnvoll, frisch gepresste hausgemachte Säfte, Smoothies und Häppchen zu verwenden. Den Gerichten sollten Quellen für Omega-3-Fettsäuren wie Fischöl, Leinsamenöl oder Leinsamen zugesetzt werden.
Es ist besser, Zucker und Süßigkeiten ganz zu vermeiden. Ein Löffel Honig mit einer Tasse Wasser schadet jedoch nicht: Bienenprodukte haben eine ausgeprägte entzündungshemmende, antioxidative und antitumorale Wirkung. Die einzige Kontraindikation für die Verwendung von Honig ist eine Allergie gegen das Produkt.
Von der Diät sollten ausgeschlossen werden:
- Fleisch, Schmalz, Innereien;
- Butter, fetthaltige Milchprodukte;
- Geräuchertes Fleisch, Wurst, Fleisch- und Fischkonserven;
- Alkohol in jeglicher Form;
- Süßigkeiten, Gebäck, Kuchen und Gebäck, Bonbons und Schokolade;
- Fertiggerichte, Fast Food, Snacks;
- Frittierte Speisen.
Sie sollten täglich ausreichend Gemüse, Grünzeug und Obst zu sich nehmen und sauberes Trinkwasser zu sich nehmen.
Während der Chemotherapie und einige Zeit danach sollten Sie selbstgemachte Gemüse- und Fruchtsäfte trinken und selbstgemachten fettarmen Hüttenkäse, Milch und Käse essen. Es ist wichtig, viel zu trinken, die Zähne zu putzen und den Mund häufig (etwa viermal täglich) auszuspülen.
Optimale Mahlzeiten für Patienten mit Hirngliom:
- Gemüseaufläufe;
- Beilagen und Suppen aus Getreide (vorzugsweise Buchweizen, Haferflocken, Reis, Couscous, Bulgur);
- Gedämpfte Käsekuchen, Puddings, Aufläufe;
- Geschmortes und gebackenes Gemüse;
- Eintöpfe, Gemüsesuppen, erste und zweite Gänge aus Hülsenfrüchten (einschließlich Soja), Pasteten und Soufflés;
- Smoothies, grüner Tee, Kompotte und Häppchen.
Verhütung
Wenn eine Person einen gesunden Lebensstil führt und es in ihrer Verwandtschaft keine Fälle von Krebserkrankungen gab, hat sie alle Chancen, kein Hirngliom zu bekommen. Es gibt keine spezifische Prävention solcher Tumoren, daher gelten die wichtigsten Präventionsmaßnahmen als richtige Ernährung, körperliche Aktivität, Vermeidung von schlechten Gewohnheiten und das Fehlen von Berufs- und Haushaltsgefahren.
Spezialisten geben eine Reihe einfacher, aber wirksamer Empfehlungen:
- Trinken Sie mehr reines Wasser und vermeiden Sie gesüßte Limonaden, abgepackte Säfte, Energydrinks und Alkohol.
- Vermeiden Sie Gefahren am Arbeitsplatz und im Haushalt: weniger Kontakt mit Chemikalien, ätzenden Lösungen und Flüssigkeiten.
- Versuchen Sie, Speisen durch Kochen, Dünsten und Backen zuzubereiten, aber nicht durch Braten. Bevorzugen Sie gesunde, hochwertige hausgemachte Lebensmittel.
- Ein großer Teil Ihrer Ernährung sollte aus pflanzlichen Lebensmitteln, einschließlich Grünzeug, bestehen, unabhängig von der Jahreszeit.
- Ein weiterer negativer Faktor ist Übergewicht, das es zu reduzieren gilt. Gewichtskontrolle ist für die Gesundheit des gesamten Körpers sehr wichtig.
- Pflanzenöle sollten Butter und Schmalz immer vorgezogen werden.
- Wenn möglich, ist es wünschenswert, umweltfreundlichen Produkten, Fleisch ohne Hormone, Gemüse und Obst ohne Nitrate und Pestizide den Vorzug zu geben. Es ist besser, rotes Fleisch ganz zu vermeiden.
- Nehmen Sie Multivitaminpräparate nicht ohne Indikation und in großen Mengen ein. Nehmen Sie keine Medikamente ohne ärztliche Verschreibung ein: Selbstmedikation ist oft sehr, sehr gefährlich.
- Wenn verdächtige Symptome auftreten, ist ein Arztbesuch erforderlich, ohne auf eine Verschlimmerung der Situation, das Auftreten von Nebenwirkungen und Komplikationen zu warten.
- Süßigkeiten und Lebensmittel mit einem hohen glykämischen Index sind ein unerwünschter Bestandteil der Ernährung.
- Je früher ein Mensch zum Arzt geht, desto besser sind seine Heilungschancen (und das gilt für fast jede Krankheit, auch für Gliome des Gehirns).
Um die Entstehung von Onkopathologien zu verhindern, ist ausreichend Schlaf und Ruhe erforderlich, übermäßiger Alkoholkonsum muss vermieden werden, hochwertige natürliche Lebensmittel müssen bevorzugt werden und die Nutzung von Geräten (insbesondere Mobiltelefonen) muss eingeschränkt werden.
Tumorerkrankungen treten häufig bei älteren und alten Menschen auf. Daher ist es wichtig, bereits in jungen Jahren auf die eigene Gesundheit zu achten und pathologische Prozesse nicht durch einen ungesunden Lebensstil und ungesunde Gewohnheiten zu provozieren.
Die genauen Ursachen der Onkologie sind noch nicht geklärt. Eine gewisse Rolle spielen jedoch natürlich ungünstige Arbeits- und Umweltbedingungen, die Belastung durch ionisierende und elektromagnetische Strahlung sowie hormonelle Veränderungen. Vermeiden Sie lange und regelmäßige Sonneneinstrahlung, plötzliche Temperaturschwankungen, Überhitzung im Bad oder in der Sauna sowie häufiges heißes Baden oder Duschen.
Eine weitere Frage: Wie kann ein Wiederauftreten eines Hirnglioms nach erfolgreicher Behandlung verhindert werden? Das Wiederauftreten von Neoplasmen ist eine komplexe und leider häufige Komplikation, die im Voraus schwer vorherzusagen ist. Patienten wird empfohlen, sich regelmäßigen Vorsorgeuntersuchungen und Kontrolluntersuchungen zu unterziehen, mindestens zweimal jährlich einen Onkologen und einen behandelnden Arzt aufzusuchen, einen gesunden Lebensstil zu führen, sich gesund und natürlich zu ernähren und moderate körperliche Aktivität auszuüben. Eine weitere Voraussetzung ist Lebensfreude, gesunder Optimismus und eine positive Einstellung zum Erfolg unter allen Umständen. Dazu gehören auch eine freundliche Atmosphäre in der Familie und am Arbeitsplatz, Geduld und bedingungslose Unterstützung durch nahestehende Menschen.
Prognose
Der Zustand des Gehirns und die Eigenschaften des Glioms zum Zeitpunkt seiner Entdeckung beeinflussen die Überlebensrate ebenso stark wie die durchgeführte Behandlung. Ein zufriedenstellender Allgemeinzustand des Patienten und sein Alter verbessern die Prognose (bei jungen Patienten ist die Prognose optimistischer). Ein wichtiger Indikator ist das histologische Bild des Neoplasmas. So haben niedriggradige Gliome eine bessere Prognose als anaplastische Gliome und insbesondere Glioblastome (die ungünstigsten Tumorprozesse). Astrozytome haben eine schlechtere Prognose als Oligodendrogliome.
Maligne Astrozytome sprechen schlecht auf die Therapie an und haben eine relativ niedrige Überlebensrate von sechs bis fünf Jahren. Gleichzeitig wird die Lebenserwartung bei niedriggradigen Gliomen auf 1-10 Jahre geschätzt.
Maligne Astrozytome sind grundsätzlich unheilbar. Die Behandlung zielt in der Regel darauf ab, neurologische Manifestationen (einschließlich kognitiver Dysfunktion) zu reduzieren und die Lebenserwartung bei gleichzeitiger Aufrechterhaltung der höchstmöglichen Lebensqualität zu erhöhen. Die symptomatische Therapie wird vor dem Hintergrund von Rehabilitationsmaßnahmen durchgeführt. Auch die Mitarbeit eines Psychologen ist wichtig.
In den letzten zehn Jahren haben Wissenschaftler einige Fortschritte im Verständnis der Natur von Hirntumoren und ihrer Behandlung erzielt. Es besteht noch viel Handlungsbedarf, um die Prognose der Erkrankung zu optimieren. Die Hauptaufgabe der Spezialisten besteht heute darin, Hirngliome sowohl in frühen als auch in späteren Entwicklungsstadien mit mehreren Maßnahmen wirksam zu behandeln.