Neue Veröffentlichungen
Neue Erkenntnisse tragen zu einem besseren Verständnis der Ursachen des Rett-Syndroms bei
Zuletzt überprüft: 02.07.2025

Alle iLive-Inhalte werden medizinisch überprüft oder auf ihre Richtigkeit überprüft.
Wir haben strenge Beschaffungsrichtlinien und verlinken nur zu seriösen Medienseiten, akademischen Forschungseinrichtungen und, wenn möglich, medizinisch begutachteten Studien. Beachten Sie, dass die Zahlen in Klammern ([1], [2] usw.) anklickbare Links zu diesen Studien sind.
Wenn Sie der Meinung sind, dass einer unserer Inhalte ungenau, veraltet oder auf andere Weise bedenklich ist, wählen Sie ihn aus und drücken Sie Strg + Eingabe.

Das Rett-Syndrom ist eine seltene neurologische Entwicklungsstörung, für die es derzeit weder eine Heilung noch eine wirksame Behandlung gibt. Es verursacht schwere körperliche und kognitive Symptome, die sich häufig mit Autismus-Spektrum-Störungen überschneiden.
Das Rett-Syndrom wird durch Mutationen im MECP2-Gen verursacht, das im Gehirn stark exprimiert wird und eine wichtige Rolle bei der Erhaltung der Gesundheit von Neuronen zu spielen scheint. Das Gen befindet sich auf dem X-Chromosom und betrifft vor allem Mädchen. Um Therapien für das Rett-Syndrom zu entwickeln, wollen Forscher MECP2 und seine Funktionen im Gehirn besser verstehen.
Forscher, darunter Rudolf Jaenisch, Mitbegründer des Whitehead Institute, untersuchen MECP2 seit Jahrzehnten, doch viele grundlegende Fakten über das Gen blieben unbekannt. Das vom Gen kodierte Protein MECP2 ist an der Genregulation beteiligt; es bindet an die DNA und beeinflusst die Expressionsstärke verschiedener anderer Gene bzw. die von ihnen produzierte Proteinmenge.
Den Forschern lag jedoch keine vollständige Liste der von MECP2 betroffenen Gene vor und es bestand kein Konsens darüber, wie MECP2 diese Gene beeinflusst.
Frühe Studien zu MECP2 deuteten darauf hin, dass es ein Repressor ist, der die Expression seiner Zielgene reduziert. Untersuchungen von Jaenisch und anderen zeigten jedoch bereits, dass MECP2 auch als Aktivator wirkt und die Expression seiner Zielgene erhöht – und dass es möglicherweise überhaupt ein Aktivator ist. Unbekannt war auch der Wirkmechanismus von MECP2, also was genau das Protein bewirkt, um Veränderungen in der Genexpression zu verursachen.
Technologische Einschränkungen haben Forscher daran gehindert, Klarheit über diese Fragen zu gewinnen. Doch Yanish, der Postdoc seines Labors Yi Liu und sein ehemaliges Labormitglied Anthony Flamier, heute Assistenzprofessor am Forschungszentrum CHU Sainte-Justine der Université de Montréal, haben mithilfe modernster Techniken diese verbleibenden Fragen zu MECP2 beantwortet und neue Erkenntnisse über seine Rolle bei der Gesundheit und Erkrankung des Gehirns gewonnen.
Ihre Ergebnisse wurden in der Zeitschrift Neuron veröffentlicht und die Forscher erstellten außerdem ein Online-Repository ihrer MECP2-Daten, das MECP2-NeuroAtlas-Portal, als Ressource für andere Forscher.
„Ich denke, diese Arbeit wird das Verständnis der Menschen darüber, wie MECP2 das Rett-Syndrom verursacht, grundlegend verändern. Wir haben ein völlig neues Verständnis des Mechanismus und es könnte neue Wege für die Entwicklung von Behandlungen für die Krankheit eröffnen“, sagt Janisch, der auch Professor für Biologie am MIT ist.
Tieferes Verständnis von MECP2 im Gehirn
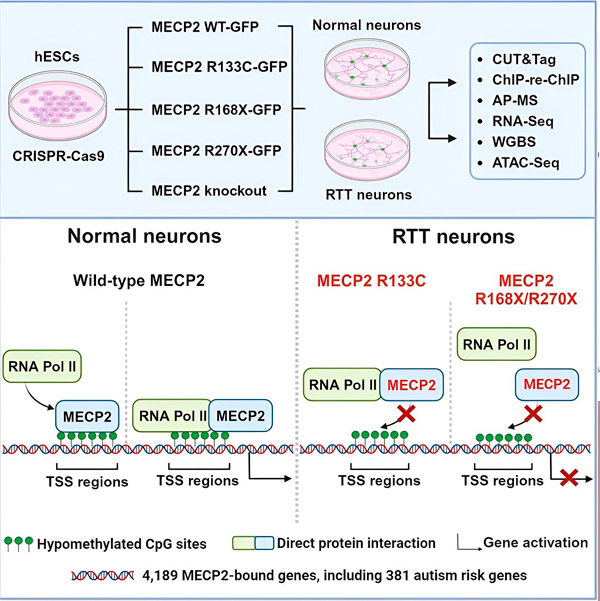
Die Forscher erstellten zunächst eine detaillierte Karte der Bindungsorte von MECP2 in menschlichen neuronalen Gensequenzen, entweder innerhalb der Gene oder in regulatorischen DNA-Regionen in deren Nähe. Sie verwendeten dafür einen Ansatz namens CUT&Tag, der Proteininteraktionen mit DNA mit hoher Präzision lokalisieren kann.
Die Forscher entdeckten mehr als 4.000 Gene, die mit MECP2 assoziiert sind. Sie wiederholten ihre Kartierung in Neuronen mit häufigen MECP2-Mutationen, die mit dem Rett-Syndrom assoziiert sind, um festzustellen, wo MECP2 im Krankheitszustand dezimiert ist.
Das Wissen, an welche Gene MECP2 bindet, ermöglichte es Liu und Flamier, Verbindungen zwischen den Zielmolekülen von MECP2 und der Gehirngesundheit herzustellen. Sie fanden heraus, dass viele seiner Zielmoleküle an der Entwicklung und Funktion neuronaler Axone und Synapsen beteiligt sind.
Sie verglichen ihre Liste der MECP2-Ziele auch mit der Datenbank der Autismus-assoziierten Gene der Simons Foundation Autism Research Initiative (SFARI) und stellten fest, dass 381 Gene in dieser Datenbank MECP2-Ziele sind.
Quelle: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Diese Erkenntnisse können dazu beitragen, die Mechanismen aufzuklären, die den Autismussymptomen beim Rett-Syndrom zugrunde liegen, und bieten einen guten Ausgangspunkt für die Untersuchung der möglichen Rolle von MECP2 bei Autismus.
„Wir haben die erste integrierte Karte des MECP2-Epigenoms im gesunden und kranken Zustand erstellt, und diese Karte kann zukünftige Forschungen leiten“, sagt Liu. „Das Wissen, welche Gene Ziele von MECP2 sind und welche Gene bei der Krankheit direkt gestört sind, bietet eine solide Grundlage für das Verständnis des Rett-Syndroms und für Fragen zur Genregulation in Neuronen.“
Die Forscher untersuchten auch, ob MECP2 die Expression seiner Zielgene erhöhte oder verringerte. In Übereinstimmung mit der Geschichte von MECP2, das von einigen als Aktivator und von anderen als Repressor bezeichnet wurde, fanden Liu und Flamier Beispiele, in denen MECP2 beide Rollen spielte.
Während MECP2 eher als Repressor angesehen wird, fanden Liu und Flamier heraus, dass es hauptsächlich ein Aktivator ist – was frühere Ergebnisse von Jaenisch und Liu bestätigte. Ein neues Experiment zeigte, dass MECP2 mindestens 80 % seiner Ziele aktiviert, ein anderes zeigte, dass es bis zu 88 % seiner Ziele aktiviert.
Die von den Forschern erstellte Karte der Zielgene lieferte zusätzliche Einblicke in die Rolle von MECP2 als Aktivator. Sie fanden heraus, dass MECP2 bei aktivierten Genen typischerweise an eine DNA-Region oberhalb des Gens bindet, die als Transkriptionsstartstelle bezeichnet wird.
Hier leitet die zelluläre Maschinerie die Transkription eines Gens in RNA ein. Anschließend wird die RNA in ein funktionelles Protein übersetzt, das das Produkt der Genexpression ist. Das Vorhandensein von MECP2 am Transkriptionsstartpunkt, wo die Genexpression beginnt, steht im Einklang mit seiner Rolle als Genaktivator.
Die Forscher untersuchten anschließend, welche Rolle MECP2 bei der Genaktivierung spielt. Sie untersuchten, an welche Moleküle MECP2 neben DNA an dieser Stelle bindet, und fanden heraus, dass MECP2 direkt mit einem Proteinkomplex namens RNA-Polymerase II (RNA-Pol II) interagiert. RNA-Pol II ist eine wichtige zelluläre Maschine, die DNA in RNA umschreibt. RNA-Pol II kann Gene nicht selbst finden und benötigt daher verschiedene Kofaktoren, sogenannte Proteinkollaborateure, die seine Aufgabe unterstützen.
Die Forscher vermuten, dass MECP2 als ein solcher Kofaktor fungiert und RNA-Pol II dabei unterstützt, die Transkription an Genen zu initiieren, an die MECP2 bindet. Strukturanalysen von MECP2 identifizierten Teile des Moleküls, die an RNA-Pol II binden. Weitere Experimente bestätigten, dass der Verlust von MECP2 die Präsenz von RNA-Pol II an geeigneten Transkriptionsstartstellen sowie die Expressionsniveaus der Zielgene verringert.
Dies deutet darauf hin, dass das Rett-Syndrom durch eine verminderte Transkription von Genen verursacht werden könnte, die von MECP2 angesteuert werden. Grund dafür sind MECP2-Mutationen, die die Bindung an RNA-Pol II oder an DNA verhindern. Im Einklang mit dieser Annahme handelt es sich bei den häufigsten mit der Krankheit assoziierten MECP2-Mutationen um Trunkierungen: Mutationen, bei denen ein Teil des Proteins fehlt, was die Interaktion zwischen MECP2 und RNA-Pol II verändern kann.
Die Forscher hoffen, dass ihre Erkenntnisse nicht nur unser Verständnis von MECP2 verändern werden, sondern dass ein tieferes und umfassenderes Verständnis der Art und Weise, wie MECP2 die Entwicklung und Funktion des Gehirns beeinflusst, zu neuen Erkenntnissen führen könnte, die Menschen mit Rett-Syndrom und verwandten Erkrankungen, einschließlich Autismus, helfen werden.
„Dieses Projekt ist ein hervorragendes Beispiel für die Zusammenarbeit im Janisch-Labor“, sagt Flamier. „Rudolf und ich hatten ein spezifisches Problem im Zusammenhang mit dem Rett-Syndrom, und ich hatte Erfahrung mit der CUT&Tag-Technologie, die das Problem lösen könnte. Im Gespräch erkannten wir, dass wir unsere Anstrengungen bündeln könnten, und verfügen nun über eine umfangreiche Informationssammlung zu MECP2 und seinen Zusammenhängen mit Krankheiten.“